 |
Главная |


Глава 2. Экспериментальные результаты и их обсуждение.
|
из
5.00
|
В данной главе будет рассмотрено несколько конкретных примеров применения программы Gaussian03 для расчета структуры и свойств молекулярных систем.
Одной из задач квантовой химии является расчет поверхности потенциальной энергии (ППЭ) молекулярных систем. ППЭ – непрерывная функция потенциальной энергии молекулярной системы от всех независимых ядерных координат. В качестве ядерных координат обычно используют внутренние координаты молекулы: длины связей, валентные и двугранные углы. Общее число независимых координат молекулы равно 3N-6(3N-5 для линейной молекулы), где N – число ядер. Для двухатомной молекулы A-B потенциальная энергия зависит от одной ядерной координаты – межъядерное расстояние r, и ППЭ имеет вид показанный на рис. 1.
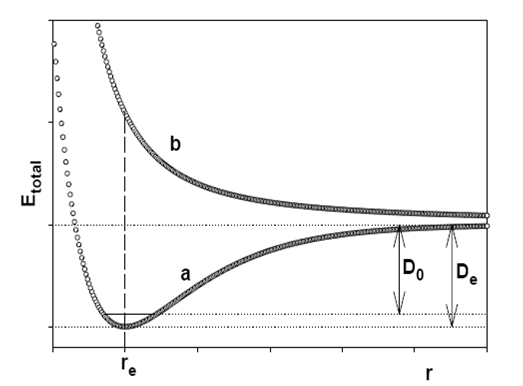
Рис.1. Типичный вид ППЭ двухатомной молекулы.
На ППЭ имеется глобальный минимум при r = re. Эта точка соответствует устойчивой геометрической конфигурации молекулы. При использовании программы Gaussian03 процедура поиска минимума на ППЭ называется оптимизацией геометрии (Geometry Optimization). Рассмотрим пример оптимизации геометрии молекулы H2.
Для проведения расчета необходимо подготовить входное задание (input-file), в котором следует указать:
· метод расчета;
· стартовую геометрию молекулы;
· некоторые другие сведения, необходимые программе для проведения расчетов.
Ниже приведен пример входного задания для программы Gaussian03.
#p B3LYP/6-311++g** opt (метод расчета)
Hydrogen (DFT) (заглавие)
0 1 (заряд и мультиплетность)
H (начальная геометрия молекулы r=1,0Å)
H 1 1.0
Используя это входное задание, программа осуществляет оптимизацию геометрию по следующему алгоритму:
· рассчитывается значение энергии и градиента при r=1,0A;
· если gradE>0, то задается новое значение r, меньше r=1,0A, и наоборот если gradE<0, то задается r>1,0A.
· процедура повторяется до тех пор, пока не будут достигнуты критерии сходимости.
Результаты вычислений проведенных программой содержаться в out-file. Часть out-file представлена ниже.
----------------------------
! Initial Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 1.0 estimate D2E/DX2 !
--------------------------------------------------------------------------------
Trust Radius=3.00D-01 FncErr=1.00D-07 GrdErr=1.00D-06
Number of steps in this run= 20 maximum allowed number of steps= 100.
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
Leave Link 103 at Thu Dec 17 15:23:21 2009, MaxMem= 6291456 cpu: 0.0
(Enter C:\G03W\l202.exe)
Input orientation:
---------------------------------------------------------------------
Center Atomic Atomic Coordinates (Angstroms)
Number Number Type X Y Z
---------------------------------------------------------------------
1 1 0 0.000000 0.000000 0.000000
2 1 0 0.000000 0.000000 1.000000
---------------------------------------------------------------------
Stoichiometry H2
Framework group D*H[C*(H.H)]
Deg. of freedom 1
Full point group D*H NOp 8
Largest Abelian subgroup D2H NOp 8
Largest concise Abelian subgroup C2 NOp 2
Standard orientation:
---------------------------------------------------------------------
Center Atomic Atomic Coordinates (Angstroms)
Number Number Type X Y Z
---------------------------------------------------------------------
1 1 0 0.000000 0.000000 0.500000
2 1 0 0.000000 0.000000 -0.500000
---------------------------------------------------------------------
Rotational constants (GHZ): 0.0000000 1002.9102017 1002.9102017
Leave Link 202 at Thu Dec 17 15:23:22 2009, MaxMem= 6291456 cpu: 0.0
(Enter C:\G03W\l301.exe)
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 0.7442 -DE/DX = -0.0001 !
--------------------------------------------------------------------------------
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
Largest change from initial coordinates is atom 1 0.128 Angstoms.
Leave Link 103 at Thu Dec 17 15:23:40 2009, MaxMem= 6291456 cpu: 0.0
(Enter C:\G03W\l202.exe)
Input orientation:
---------------------------------------------------------------------
Center Atomic Atomic Coordinates (Angstroms)
Number Number Type X Y Z
---------------------------------------------------------------------
1 1 0 0.000000 0.000000 0.127881
2 1 0 0.000000 0.000000 0.872119
---------------------------------------------------------------------
Stoichiometry H2
Framework group D*H[C*(H.H)]
Deg. of freedom 1
Full point group D*H NOp 8
Largest Abelian subgroup D2H NOp 8
Largest concise Abelian subgroup C2 NOp 2
Standard orientation:
---------------------------------------------------------------------
Center Atomic Atomic Coordinates (Angstroms)
Number Number Type X Y Z
---------------------------------------------------------------------
1 1 0 0.000000 0.000000 0.372119
2 1 0 0.000000 0.000000 -0.372119
---------------------------------------------------------------------
Rotational constants (GHZ): 0.0000000 1810.6704785 1810.6704785
Leave Link 202 at Thu Dec 17 15:23:41 2009, MaxMem= 6291456 cpu: 1.0
(Enter C:\G03W\l601.exe)
Copying SCF densities to generalized density rwf, ISCF=0 IROHF=0.
Результаты вычислений представлены в таблице 1.
Таблица 1. Результаты оптимизации герметрии молекулы водорода с использованием программы Gaussian03.
| r, Å | E, ккал/моль | gradE, a. e. |
| 1,0000 0,8412 0,7367 0,7442 | -723,387 -736,976 -740,181 -740,205 | 0,08677 0,05082 -0,00535 0,00006 |
Из полученных данных видно, что для оптимизации геометрии программе потребовалось провести расчет всего 4 точек на ППЭ. Полученное значение длины связи r=0,7442Å соответствует литературному[].
Программы для квантово-химических расчетов позволяют вычислять большое число характеристик. Почти любой расчет включает оптимизацию геометрии молекулы. При этом на каждом шаге вычисляется полная энергия и градиент. На последнем шаге такого расчета помимо значений полной энергии автоматически можно получить информацию о равновесной геометрии молекулы (длины связей, валентные углы и т. д.). Для установления типа стационарной точки проводят расчет колебательного спектра, который требует больших затрат компьютерного времени, но в результате кроме колебательных частот автоматически вычисляются:
· декартовы смещения атомов, соответствующие данному колебанию;
· интенсивности колебаний в ИК-спектре;
· изменение термодинамических функций и теплоемкости в интервале от 0 до Т для вещества, находящегося в состоянии идеального газа.
В качестве примера рассмотрим фрагмент файла оптимизации геометрии и расчета колебательного спектра формальдегида.
Входное задание выглядит следующим образом:
%chk=ch2o
#RHF/sto-3G opt pop=full freq
formaldehyde
0 1
c
o 1 1.2
h 1 1.08 2 122.5
h 1 1.08 2 122.5 3 180.0
Фрагмент out-file:
----------------------------
! Initial Parameters !
{стартовая геометрия }
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 1.2 estimate D2E/DX2 !
! R2 R(1,3) 1.08 estimate D2E/DX2 !
! R3 R(1,4) 1.08 estimate D2E/DX2 !
! A1 A(2,1,3) 122.5 estimate D2E/DX2 !
! A2 A(2,1,4) 122.5 estimate D2E/DX2 !
! A3 A(3,1,4) 115.0 estimate D2E/DX2 !
! A4 L(2,1,3,4,-2) 180.0 estimate D2E/DX2 !
--------------------------------------------------------------------------------
Distance matrix (angstroms):
1 2 3 4
1 C 0.000000
2 O 1.200000 0.000000
3 H 1.080000 1.999770 0.000000
4 H 1.080000 1.999770 1.821726 0.000000
Stoichiometry CH2O { симметрия молекулы }
Framework group C2V[C2(CO),SGV(H2)]
Deg. of freedom 3
Full point group C2V NOp 4
Largest Abelian subgroup C2V NOp 4
Largest concise Abelian subgroup C2 NOp 2
Standard orientation:
---------------------------------------------------------------------
Center Atomic Atomic Coordinates (Angstroms)
Number Number Type X Y Z
---------------------------------------------------------------------
1 6 0 0.000000 0.000000 -0.527465
2 8 0 0.000000 0.000000 0.672535
3 1 0 0.000000 0.910863 -1.107748
4 1 0 0.000000 -0.910863 -1.107748
---------------------------------------------------------------------
Rotational constants (GHZ): 302.2011851 39.2189297 34.7138511
Standard basis: STO-3G (5D, 7F)
There are 7 symmetry adapted basis functions of A1 symmetry.
There are 0 symmetry adapted basis functions of A2 symmetry.
There are 2 symmetry adapted basis functions of B1 symmetry.
There are 3 symmetry adapted basis functions of B2 symmetry.
Initial guess orbital symmetries: {симметрия начальных МО }
Occupied (A1) (A1) (A1) (A1) (B2) (A1) (B1) (B2)
Virtual (B1) (A1) (B2) (A1)
Closed chell SCF: { решения уравнения Рутаана }
Requested convergence on RMS density matrix=1.00D-08 within 128 cycles.
Requested convergence on MAX density matrix=1.00D-06.
SCF Done: E(RHF) = -112.352904580 {полная энергия } A.U. after 9 cycles
Convg = 0.7537D-08 -V/T = 2.0084
S**2 = 0.0000
Compute integral first derivatives. {вычисление градиента энергии }
-------------------------------------------------------------------
Center Atomic Forces (Hartrees/Bohr)
Number Number X Y Z
-------------------------------------------------------------------
1 6 0.000000000 0.000000000 -0.016779441
2 8 0.000000000 0.000000000 0.041072257
3 1 0.015861411 0.000000000 -0.012146408
4 1 -0.015861411 0.000000000 -0.012146408
-------------------------------------------------------------------
Cartesian Forces: Max 0.041072257 RMS 0.015184200
Internal Forces: Max 0.041072257 RMS 0.018850815
Variable Old X -DE/DX Delta X Delta X Delta X New X
(Linear) (Quad) (Total)
R1 2.26767 0.04107 0.00000 0.03904 0.03904 2.30671
R2 2.04090 0.01990 0.00000 0.05471 0.05471 2.09562
R3 2.04090 0.01990 0.00000 0.05471 0.05471 2.09562
A1 2.13803 0.00117 0.00000 0.00715 0.00715 2.14518
A2 2.13803 0.00117 0.00000 0.00715 0.00715 2.14518
A3 2.00713 -0.00234 0.00000 -0.01430 -0.01430 1.99283
A4 3.14159 0.00000 0.00000 0.00000 0.00000 3.14159
Item Value Threshold Converged?
Maximum Force 0.041072 0.000450 NO
RMS Force 0.018851 0.000300 NO
Maximum Displacement 0.050283 0.001800 NO
RMS Displacement 0.032608 0.001200 NO
Predicted change in Energy=-1.930840D-03 {критерии сходимости не достигнуты, поэтому вычисляют новые значения ядерных координат и приступают ко второму циклу оптимизации}
Distance matrix (angstroms): {второй цикл оптимизации}
1 2 3 4
1 C 0.000000
2 O 1.220659 0.000000
3 H 1.108953 2.047122 0.000000
4 H 1.108953 2.047122 1.861996 0.000000
Stoichiometry CH2O
Framework group C2V[C2(CO),SGV(H2)]
Deg. of freedom 3
Full point group C2V NOp 4
Largest Abelian subgroup C2V NOp 4
Largest concise Abelian subgroup C2 NOp 2
Standard orientation: { новая геометрия }
---------------------------------------------------------------------
Center Atomic Atomic Coordinates (Angstroms)
Number Number Type X Y Z
---------------------------------------------------------------------
1 6 0 0.000000 0.000000 -0.535015
2 8 0 0.000000 0.000000 0.685643
3 1 0 0.000000 0.930998 -1.137528
4 1 0 0.000000 -0.930998 -1.137528
Closed shell SCF:
Requested convergence on RMS density matrix=1.00D-08 within 128 cycles.
Requested convergence on MAX density matrix=1.00D-06.
SCF Done: E(RHF) = -112.354213361 {новое значение полной энергии} A.U. after 9 cycles
Convg = 0.1073D-08 -V/T = 2.0091
S**2 = 0.0000
Compute integral first derivatives.
-------------------------------------------------------------------
Center Atomic Forces (Hartrees/Bohr)
Number Number X Y Z
-------------------------------------------------------------------
1 6 0.000000000 0.000000000 0.001181143
2 8 0.000000000 0.000000000 -0.009586699
3 1 -0.004946323 0.000000000 0.004202778
4 1 0.004946323 0.000000000 0.004202778
-------------------------------------------------------------------
Cartesian Forces: Max 0.009586699 RMS 0.003846631
Internal Forces: Max 0.009586699 RMS 0.005025930 {новое значение градиента }
Variable Old X -DE/DX Delta X Delta X Delta X New X
(Linear) (Quad) (Total)
R1 2.30671 -0.00959 -0.00920 0.00204 -0.00716 2.29955
R2 2.09562 -0.00644 -0.01289 -0.00168 -0.01457 2.08105
R3 2.09562 -0.00644 -0.01289 -0.00168 -0.01457 2.08105
A1 2.14518 -0.00059 -0.00168 -0.00141 -0.00309 2.14209
A2 2.14518 -0.00059 -0.00168 -0.00141 -0.00309 2.14209
A3 1.99283 0.00117 0.00337 0.00281 0.00618 1.99901
A4 3.14159 0.00000 0.00000 0.00000 0.00000 3.14159
Item Value Threshold Converged?
Maximum Force 0.009587 0.000450 NO
RMS Force 0.005026 0.000300 NO
Maximum Displacement 0.012032 0.001800 NO
RMS Displacement 0.008145 0.001200 NO
Predicted change in Energy=-1.259490D-04 {критерии сходимости не достигнуты, поэтому вычисляют новые значения ядерных координат и приступают к следующему циклу оптимизации}
{ НЕСКОЛЬКО ЦИКЛОВ ОПТИМИЗАЦИИ }
Compute integral first derivatives.
-------------------------------------------------------------------
Center Atomic Forces (Hartrees/Bohr)
Number Number X Y Z
-------------------------------------------------------------------
1 6 0.000000000 0.000000000 0.000434399
2 8 0.000000000 0.000000000 -0.000298069
3 1 0.000086262 0.000000000 -0.000068165
4 1 -0.000086262 0.000000000 -0.000068165
-------------------------------------------------------------------
Cartesian Forces: Max 0.000434399 RMS 0.000158567
Internal Forces: Max 0.000298069 RMS 0.000127123
Variable Old X -DE/DX Delta X Delta X Delta X New X
(Linear) (Quad) (Total)
R1 2.29955 -0.00030 0.00003 -0.00033 -0.00029 2.29926
R2 2.08105 0.00011 0.00006 0.00022 0.00029 2.08133
R3 2.08105 0.00011 0.00006 0.00022 0.00029 2.08133
A1 2.14209 0.00001 0.00001 0.00003 0.00004 2.14213
A2 2.14209 0.00001 0.00001 0.00003 0.00004 2.14213
A3 1.99901 -0.00001 -0.00003 -0.00006 -0.00009 1.99892
A4 3.14159 0.00000 0.00000 0.00000 0.00000 3.14159
Item Value Threshold Converged?
Maximum Force 0.000298 0.000450 YES
RMS Force 0.000127 0.000300 YES
Maximum Displacement 0.000191 0.001800 YES
RMS Displacement 0.000133 0.001200 YES
Predicted change in Energy=-7.640079D-08
Optimization completed. {оптимизация геометрии завершена }
Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 1.2169 -DE/DX = -0.0003 !
! R2 R(1,3) 1.1012 -DE/DX = 0.0001 !
! R3 R(1,4) 1.1012 -DE/DX = 0.0001 !
! A1 A(2,1,3) 122.7326 -DE/DX = 0.0 !
! A2 A(2,1,4) 122.7326 -DE/DX = 0.0 !
! A3 A(3,1,4) 114.5348 -DE/DX = 0.0 !
! A4 L(2,1,3,4,-2) 180.0 -DE/DX = 0.0 !
--------------------------------------------------------------------------------
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
**********************************************************************
Population analysis using the SCF density.
**********************************************************************
Orbital symmetries:
Occupied (A1) (A1) (A1) (A1) (B2) (A1) (B1) (B2)
Virtual (B1) (A1) (B2) (A1)
The electronic state is 1-A1.
Alpha occ. eigenvalues -- -20.31268 -11.12503 -1.33734 -0.80781 -0.63291
Alpha occ. eigenvalues -- -0.54550 -0.44311 -0.35440
Alpha virt. eigenvalues -- 0.28193 0.62875 0.73453 0.91272
Molecular Orbital Coefficients {энергии и коэффициенты МО }
1 2 3 4 5
(A1)--O (A1)--O (A1)--O (A1)--O (B2)--O
EIGENVALUES -- -20.31268 -11.12503 -1.33734 -0.80781 -0.63291
1 1 C 1S 0.00053 0.99263 -0.12252 -0.18563 0.00000
2 2S -0.00718 0.03290 0.27717 0.57739 0.00000
3 2PX 0.00000 0.00000 0.00000 0.00000 0.00000
4 2PY 0.00000 0.00000 0.00000 0.00000 0.53320
5 2PZ -0.00629 0.00052 0.15771 -0.22622 0.00000
6 2 O 1S 0.99429 0.00013 -0.21937 0.09884 0.00000
7 2S 0.02593 -0.00571 0.76903 -0.42912 0.00000
8 2PX 0.00000 0.00000 0.00000 0.00000 0.00000
9 2PY 0.00000 0.00000 0.00000 0.00000 0.44215
10 2PZ -0.00562 0.00164 -0.17012 -0.16460 0.00000
11 3 H 1S 0.00019 -0.00651 0.03176 0.26456 0.30031
12 4 H 1S 0.00019 -0.00651 0.03176 0.26456 -0.30031
6 7 8 9 10
(A1)--O (B1)--O (B2)--O (B1)--V (A1)--V
EIGENVALUES -- -0.54550 -0.44311 -0.35440 0.28193 0.62875
1 1 C 1S 0.03302 0.00000 0.00000 0.00000 -0.20805
2 2S -0.10678 0.00000 0.00000 0.00000 1.30330
3 2PX 0.00000 0.60939 0.00000 0.82106 0.00000
4 2PY 0.00000 0.00000 -0.18202 0.00000 0.00000
5 2PZ -0.44755 0.00000 0.00000 0.00000 -0.44486
6 2 O 1S -0.09380 0.00000 0.00000 0.00000 0.02811
7 2S 0.49905 0.00000 0.00000 0.00000 -0.16153
8 2PX 0.00000 0.67586 0.00000 -0.76728 0.00000
9 2PY 0.00000 0.00000 0.87002 0.00000 0.00000
10 2PZ 0.67686 0.00000 0.00000 0.00000 0.24618
11 3 H 1S 0.15894 0.00000 -0.35908 0.00000 -0.88936
12 4 H 1S 0.15894 0.00000 0.35908 0.00000 -0.88936
11 12
(B2)--V (A1)--V
EIGENVALUES -- 0.73453 0.91272
1 1 C 1S 0.00000 -0.09474
2 2S 0.00000 0.63122
3 2PX 0.00000 0.00000
4 2PY 1.14850 0.00000
5 2PZ 0.00000 1.17306
6 2 O 1S 0.00000 0.11575
7 2S 0.00000 -0.86356
8 2PX 0.00000 0.00000
9 2PY -0.31847 0.00000
10 2PZ 0.00000 0.92383
11 3 H 1S -0.84000 0.15485
12 4 H 1S 0.84000 0.15485
DENSITY MATRIX. {матрица плотности }
1 2 3 4 5
1 1 C 1S 2.07173
2 2S -0.22403 0.84548
3 2PX 0.00000 0.00000 0.74272
4 2PY 0.00000 0.00000 0.00000 0.63487
5 2PZ 0.01680 -0.07811 0.00000 0.00000 0.55277
6 2 O 1S 0.01217 -0.00170 0.00000 0.00000 -0.04246
7 2S -0.00748 -0.17656 0.00000 0.00000 -0.01031
8 2PX 0.00000 0.00000 0.82373 0.00000 0.00000
9 2PY 0.00000 0.00000 0.00000 0.15478 0.00000
10 2PZ 0.15075 -0.42874 0.00000 0.00000 -0.58497
11 3 H 1S -0.10843 0.28874 0.00000 0.45097 -0.25195
12 4 H 1S -0.10843 0.28874 0.00000 -0.45097 -0.25195
6 7 8 9 10
6 2 O 1S 2.11060
7 2S -0.46429 2.05062
8 2PX 0.00000 0.00000 0.91358
9 2PY 0.00000 0.00000 0.00000 1.90486
10 2PZ -0.09605 0.55487 0.00000 0.00000 1.02842
11 3 H 1S 0.00892 -0.01948 0.00000 -0.35926 0.11724
12 4 H 1S 0.00892 -0.01948 0.00000 0.35926 0.11724
11 12
11 3 H 1S 0.63086
12 4 H 1S -0.24564 0.63086
Full Mulliken population analysis: {малликеновский анализ заселенностей }
1 2 3 4 5
1 1 C 1S 2.07173
2 2S -0.05564 0.84548
3 2PX 0.00000 0.00000 0.74272
4 2PY 0.00000 0.00000 0.00000 0.63487
5 2PZ 0.00000 0.00000 0.00000 0.00000 0.55277
6 2 O 1S 0.00000 -0.00006 0.00000 0.00000 -0.00260
7 2S -0.00027 -0.06375 0.00000 0.00000 -0.00454
8 2PX 0.00000 0.00000 0.17185 0.00000 0.00000
9 2PY 0.00000 0.00000 0.00000 0.03229 0.00000
10 2PZ -0.00905 0.13728 0.00000 0.00000 0.18337
11 3 H 1S -0.00658 0.13984 0.00000 0.17579 0.06313
12 4 H 1S -0.00658 0.13984 0.00000 0.17579 0.06313
6 7 8 9 10
6 2 O 1S 2.11060
7 2S -0.10990 2.05062
8 2PX 0.00000 0.00000 0.91358
9 2PY 0.00000 0.00000 0.00000 1.90486
10 2PZ 0.00000 0.00000 0.00000 0.00000 1.02842
11 3 H 1S 0.00004 -0.00137 0.00000 -0.01289 -0.00823
12 4 H 1S 0.00004 -0.00137 0.00000 -0.01289 -0.00823
11 12
11 3 H 1S 0.63086
12 4 H 1S -0.03711 0.63086
Gross orbital populations: {N(rk)}
1
1 1 C 1S 1.99361
2 2S 1.14297
3 2PX 0.91457
4 2PY 1.01874
5 2PZ 0.85526
6 2 O 1S 1.99812
7 2S 1.86941
8 2PX 1.08543
9 2PY 1.91138
10 2PZ 1.32356
11 3 H 1S 0.94348
12 4 H 1S 0.94348
Condensed to atoms (all electrons):
1 2 3 4
1 C 4.736285 0.444509 0.372179 0.372179
2 O 0.444509 7.788280 -0.022446 -0.022446
3 H 0.372179 -0.022446 0.630857 -0.037114
4 H 0.372179 -0.022446 -0.037114 0.630857
Mulliken atomic charges: { заряды на атомах }
1
1 C 0.074848
2 O -0.187897
3 H 0.056524
4 H 0.056524
Sum of Mulliken charges= 0.00000
Atomic charges with hydrogens summed into heavy atoms:
1
1 C 0.187897
2 O -0.187897
3 H 0.000000
4 H 0.000000
Sum of Mulliken charges= 0.00000
Electronic spatial extent (au): <R**2>= 58.6719
Charge= 0.0000 electrons
Dipole moment (field-independent basis, Debye): {дипольный момент }
X= 0.0000 Y= 0.0000 Z= -1.5370 Tot= 1.5370
Compute integral second derivatives. { вычисление вторых производных }
Total kinetic energy from orbitals= 1.113600771198D+02
Exact polarizability: 2.480 0.000 5.900 0.000 0.000 10.670
Approx polarizability: 1.781 0.000 4.706 0.000 0.000 12.936
Full mass-weighted force constant matrix:
Low frequencies --- -35.9443 -0.0013 -0.0009 0.0008 10.6335 17.2079
Low frequencies --- 1278.2740 1397.2432 1766.9474
Diagonal vibrational polarizability:
0.1016141 0.4473973 0.0915074
Diagonal vibrational hyperpolarizability:
0.0000000 0.0000000 -3.2122177
Harmonic frequencies (cm**-1), IR intensities (KM/Mole), Raman scattering
activities (A**4/AMU), depolarization ratios for plane and unpolarized
incident light, reduced masses (AMU), force constants (mDyne/A),
and normal coordinates:
1 2 3
B1 B2 A1
Frequencies -- 1278.2740 1397.2431 1766.9474
Red. masses -- 1.3683 1.3448 1.1636
Frc consts -- 1.3173 1.5468 2.1404
IR Inten -- 6.1698 29.5464 4.1653
Raman Activ -- 0.0076 3.0445 16.3920
Depolar (P) -- 0.7500 0.7500 0.7071
Depolar (U) -- 0.8571 0.8571 0.8284
Atom AN X Y Z X Y Z X Y Z
1 6 0.17 0.00 0.00 0.00 0.15 0.00 0.00 0.00 -0.03
2 8 -0.04 0.00 0.00 0.00 -0.08 0.00 0.00 0.00 0.10
3 1 -0.70 0.00 0.00 0.00 -0.27 -0.64 0.00 -0.36 -0.61
4 1 -0.70 0.00 0.00 0.00 -0.27 0.64 0.00 0.36 -0.61
4 5 6
A1 A1 B2
Frequencies -- 2099.0199 3499.9280 3647.0068
Red. masses -- 5.0502 1.0642 1.1152
Frc consts -- 13.1097 7.6809 8.7394
IR Inten -- 8.4411 1.8414 19.8276
Raman Activ -- 6.7109 38.6608 24.5167
Depolar (P) -- 0.0756 0.1865 0.7500
Depolar (U) -- 0.1406 0.3144 0.8571
Atom AN X Y Z X Y Z X Y Z
1 6 0.00 0.00 0.47 0.00 0.00 0.07 0.00 0.10 0.00
2 8 0.00 0.00 -0.32 0.00 0.00 -0.01 0.00 0.00 0.00
3 1 0.00 -0.52 -0.25 0.00 0.59 -0.38 0.00 -0.59 0.38
4 1 0.00 0.52 -0.25 0.00 -0.59 -0.38 0.00 -0.59 -0.38
-------------------
- Thermochemistry -
-------------------
Temperature 298.150 Kelvin. Pressure 1.00000 Atm.
Atom 1 has atomic number 6 and mass 12.00000
Atom 2 has atomic number 8 and mass 15.99491
Atom 3 has atomic number 1 and mass 1.00783
Atom 4 has atomic number 1 and mass 1.00783
Molecular mass: 30.01056 amu.
Principal axes and moments of inertia in atomic units:
1 2 3
EIGENVALUES -- 6.17704 47.44137 53.61841
X 0.00000 0.00000 1.00000
Y 0.00000 1.00000 0.00000
Z 1.00000 0.00000 0.00000
This molecule is an asymmetric top.
Rotational symmetry number 2.
Rotational temperatures (Kelvin) 14.02190 1.82570 1.61537
Rotational constants (GHZ): 292.16932 38.04150 33.65898
Zero-point vibrational energy 81874.9 (Joules/Mol)
19.56858 (Kcal/Mol)
Vibrational temperatures: 1839.15 2010.32 2542.24 3020.02 5035.61
(Kelvin) 5247.22
Zero-point correction= 0.031185 (Hartree/Particle)
Thermal correction to Energy= 0.034039
Thermal correction to Enthalpy= 0.034983
Thermal correction to Gibbs Free Energy= 0.010177
Sum of electronic and zero-point Energies= -112.323163
Sum of electronic and thermal Energies= -112.320308
Sum of electronic and thermal Enthalpies= -112.319364
Sum of electronic and thermal Free Energies= -112.344170
E (Thermal) CV S
KCal/Mol Cal/Mol-Kelvin Cal/Mol-Kelvin
Total 21.360 6.264 52.209
Electronic 0.000 0.000 0.000
Translational 0.889 2.981 36.130
Rotational 0.889 2.981 16.026
Vibrational 19.582 0.303 0.053
Q Log10(Q) Ln(Q)
Total Bot 0.208442D-04 -4.681015 -10.778436
Total V=0 0.460082D+10 9.662835 22.249500
Vib (Bot) 0.454649D-14 -14.342324 -33.024421
Vib (V=0) 0.100352D+01 0.001526 0.003515
Electronic 0.100000D+01 0.000000 0.000000
Translational 0.646199D+07 6.810366 15.681448
Rotational 0.709483D+03 2.850942 6.564537
В этом примере жирным шрифтом отмечены различные характеристики молекулы формальдегида, рассчитанные программой, такие как полная энергия, коэффициенты и энергии молекулярных орбиталей, дипольный момент, заряды на атомах и др. Полученные данные согласуются с экспериментальными, что подтверждает качество расчетов программы Gaussian03.
Заключение
Таким образом, использование программы Gaussian03 позволяет исследователю расчетным путем получить различные характеристики молекулярных систем (с определенной долей точности), которые согласуются с экспериментально получаемыми величинами. Это, с одной стороны, позволяет экономить время, затрачиваемое на исследование, с другой, дает возможность рассматривать системы с различных сторон, используя данные, которые не всегда можно получить экспериментально, что важно для понимания истинной сути происходящих процессов.
В работе показано, что Gaussian03 является мощным инструментом в руках исследователей, позволяющим решать задачи различной степени сложности.
Список литературы к реферату
1)http://www.msg.ameslab.gov/GAMESS/GAMESS.html
2)http://www.gaussian.com/
3)http://www.kjemi.uio.no/software/dalton/dalton.html
4) http://www.emsl.pnl.gov/docs/nwchem/nwchem.html
5) http://www.hyper.com/
6)http://www.qchem.ru/d/lect/khsl_qchem/20Lecture-19.pdf
7) Глинка, Н.Л. Общая химия / Н.Л. Глинка. – М.: Химия, 1978. – С. 111-153.
8) Матулис, Вадим Э. Прикладная квантовая химия / Вадим Э. Матулис, Виталий Э. Матулия, О. А. Ивашкевич. – Минск: БГУ, 2007. – 143с.
|
из
5.00
|
Обсуждение в статье: Глава 2. Экспериментальные результаты и их обсуждение. |
|
Обсуждений еще не было, будьте первым... ↓↓↓ |

Почему 1285321 студент выбрали МегаОбучалку...
Система поиска информации
Мобильная версия сайта
Удобная навигация
Нет шокирующей рекламы