 |
Главная |


Колебательная спектроскопия
|
из
5.00
|
Типы колебаний
Энергия, необходимая для возбуждения колебаний атомов в молекуле, соответствует энергии квантов света с длиной волны 1-15 мкм или волновым числом 400÷4000 см-1, т. е. электромагнитному излучению средней инфракрасной (ИК) области. Колебательные уровни молекул квантованы, энергия переходов между ними и, следовательно, частоты колебаний могут иметь только строго определенные значения. Поглощая квант света, молекула может переходить на более высокий колебательный уровень, обычно из основного колебательного состояния в возбужденное. Поглощенная энергия передается затем на возбуждение вращательных уровней или преобразуется в кинетическую энергию молекул. Колебания молекул проявляются в двух типах спектров: спектры поглощения в инфракрасной области (ИК-спектры) и спектры комбинационного рассеяния света (спектры КР).
Математическая модель колебаний многоатомных молекул сложна. Расчеты проведены только для простейших двухатомных молекул. Колебательная спектроскопия носит в основном эмпирический характер, т.е. основные частоты колебаний получены при сопоставлении спектров многих соединений одного класса. Это, однако, не умаляет ценности метода.
Основными типами колебаний являются валентные и деформационные.
Валентными колебаниями называются колебания ядер атомов вдоль линии связи, они обозначаются буквой n (nC=C, nC=O и т. д.).
Приближенной механической моделью валентных колебаний может служить система из двух шаров, связанных жесткой пружиной (здесь шары изображают атомы, а пружина - химическую связь) (см. рис., А).
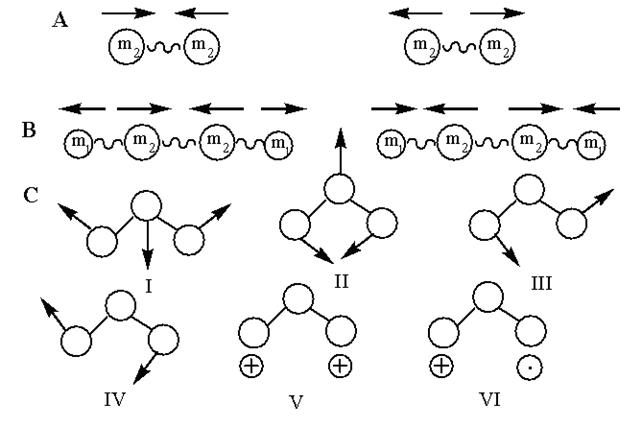
А, В – валентные колебания в молекулах;
С – деформационные колебания: I,II – ножничные; III,IV – маятниковые; V – веерные; VI – крутильные.
При растяжении или сжатии пружины шары начнут колебаться вокруг положения равновесия, т. е. будет осуществляться гармоническое колебание, описываемое уравнением
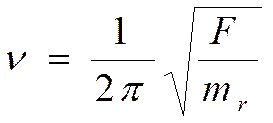
где n - частота колебания; F- силовая постоянная, характеризующая прочность связи, или силу, возвращающую шары в положение равновесия; mr - приведенная масса шаров (атомов), вычисляемая по формулам
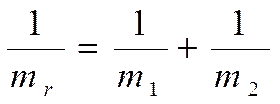
Частоты валентных колебаний определяются массой атомов и прочностью (энергией) связи. Чем масса больше, тем меньше частота, например:
nC-C » 1000 см-1; nC-Н » 3000 см-1
Чем связь прочнее, тем выше частота колебаний, например:
| nC-C »1000 см-1 | nC-О »1100 см-1 | nC-N »1050 см-1 |
| nC=C »1600 см-1 | nC=О »1700 см-1 | nC=N »1650 см-1 |
Возможно появление обертонов - колебаний, частота которых больше в целое число раз, чем у основных (2n, 3n и т. д.). Обычно интенсивность обертонов много меньше: для первого обертона она составляет 1—10 % от интенсивности основного колебания; третий обертон обнаружить обычно не удается.
В системе из трех или четырех атомов возможны два типа валентных колебаний - синфазное (в одной фазе, или симметричное, ns) и антифазное (в разных фазах, или антисимметричное, nas) (рис. В), хотя термины полностью применимы и для симметричных молекул. Частота антифазного колебания всегда выше, чем синфазного.
Деформационные колебания связаны с изменением валентного угла, образованного связями у общего атома; они обозначаются буквой d. Виды некоторых деформационных колебаний показаны на рис., С. Для возбуждения деформационных колебаний требуется меньшая энергия, чем в случае валентных колебаний, и, следовательно, они имеют меньшую частоту.
С увеличением числа атомов в молекуле число возможных колебаний быстро растет. В реальной молекуле колебания атомов тесно связаны друг с другом и взаимодействуют между собой. Спектры молекул представляют собой сложный набор различных колебаний, каждое из которых проявляется в узком интервале частот.
Интенсивность поглощения определяется, как и в УФ спектроскопии, молярным коэффициентом поглощения, однако в этом случае точность измерения существенно меньше. Обычно интенсивность полос выражают как поглощение (А) или пропускание (Т) светового потока в процентах. Полосы также оценивают по интенсивности как сильные (с.), средние (ср.) и слабые (сл.).
Получение ИК спектров
В основе получения ИК спектров лежит прямое поглощение излучения при прохождении через слой вещества. Из обширного диапазона ИК излучения обычно используют среднюю область (400- 4000 см-1). В области ближнего ИК (4000÷14300 см-1), где проявляются в основном обертоны, проводят иногда количественный анализ. В дальнюю ИК-область (100÷400 см-1) попадают практически только колебания связей углерод-металл.
Схема ИК спектрометра сходна со схемой УФ спектрометра, однако конструкция приборов более сложна. ИК излучение является тепловым; его источником обычно служит керамический стержень, раскаляемый проходящим электрическим током. С помощью системы зеркал световой поток разделяется на два одинаковых луча, один из которых пропускается через кювету с веществом, другой - через кювету сравнения. Прошедшее через кюветы излучение поступает в монохроматор, состоящий из вращающейся призмы, зеркала и щели, позволяющий выделять излучение со строго определенной частотой и плавно изменять эту частоту. Учитывая, что в ИК области большинство веществ непрозрачно, призмы изготовляют из монокристаллов солей. В приборах высокого класса применяют три призмы: из LiF (2000÷3800 см-1), NaCl(700÷2000 см-1) и КВr (400÷700 см-1). Каждая из призм в другом интервале волновых чисел дает значительно меньшее разрешение. В ряде приборов дисперсия излучения осуществляется с помощью дифракционных решеток. Интенсивности двух световых потоков (основного и луча сравнения), прошедших через монохроматор, автоматически вычитаются одна из другой. Электрический сигнал, образующийся при попадании результирующего светового потока на детектор типа термопары, усиливается и регистрируется самопишущим потенциометром. Запись представляет собой ИК спектр в виде зависимости поглощения или пропускания (в %) от частоты (в см-1) или длины волны (в мкм). Типичный вид спектра представлен на рис.

ИК спектры чаще всего получают следующим образом:
1. Растворы веществ наиболее удобны для получения спектров, так как в этом случае отсутствуют межмолекулярные взаимодействия. В связи с тем, что в ИК области поглощает любое вещество, в качестве растворителей используют соединения простейшей структуры, спектр которых имеет простейший вид (минимальное число полос), и наиболее часто - четыреххлористый углерод, который прозрачен выше 1300 см-1, а также сероуглерод, практически прозрачный и ниже 1300 см-1. Последовательно растворив вещество в том и другом растворителе, удается записать весь ИК- спектр.
Для растворов применяют цилиндрические кюветы толщиной 0,1 ÷ 1,0 мм с окнами из солевых пластин. Необходимый для заполнения кюветы объем раствора 0,1 ÷ 1,0 мл при концентрации 0,05 ÷ 10%.
2. Тонкие пленки (<0,01 мм) жидкого вещества, помещенные между солевыми пластинами, удерживаемыми капиллярными силами.
3. Пасты, приготовляемые тщательным растиранием твердого образца с вазелиновым маслом и помещаемые в виде тонкого слоя между солевыми пластинами. Само вазелиновое масло, являющееся смесью углеводородов, интенсивно поглощает в области »2900 см-1 и »1400 см-1. Иногда для приготовления паст используется гексахлорбутадиен, прозрачный выше 1600 см-1 и в области 1250÷1500 см-1, т. е. в тех интервалах частот, в которых поглощает вазелиновое масло.
4. Твердые вещества в виде тонкого порошка (0,5÷1,0 мг), тщательно перемешанные с порошком бромида калия (~100 мг) и затем спрессованные в специальном устройстве под давлением до »4,5×108 Па в тонкую пластину.
5. Метод нарушенного полного внутреннего отражения(НПВО):
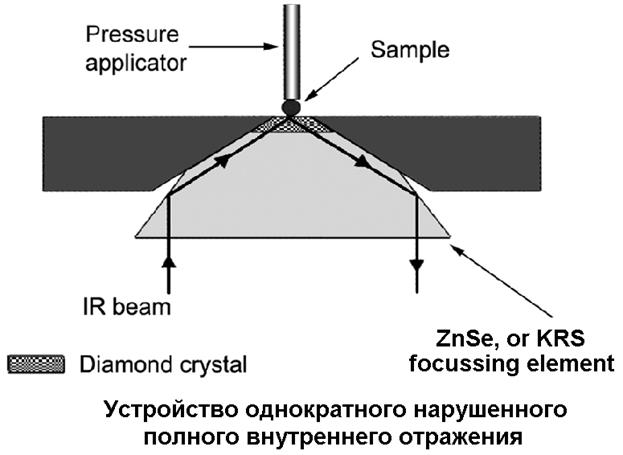
Количество вещества, необходимое для получения ИК спектра, независимо от способа приготовления пробы составляет 0,5÷2 мг.
Так как материалом для кювет являются солевые пластины, образец не должен содержать воды. Метод ИК- спектроскопии - один из наиболее доступных в лабораторной практике. Приборы просты в обращении, для получения спектра требуется всего несколько минут.
Другим типом спектров, несущим информацию о колебаниях в этом диапазоне, являются спектры комбинационного рассеяния (КР).
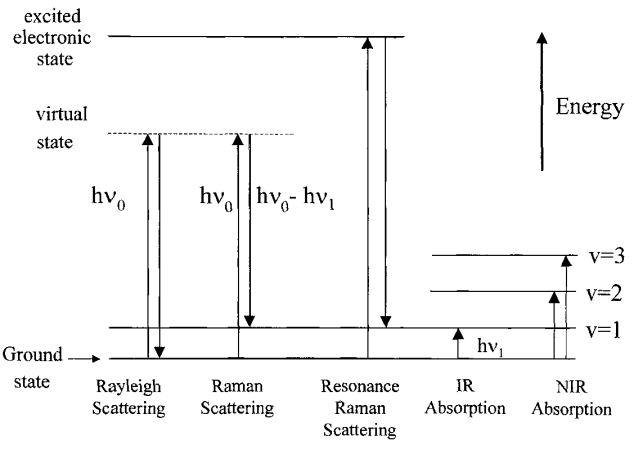
Основной особенностью их является фиксация длин волн преимущественно в видимом диапазоне. Условием их получения является наличие высокоинтенсивного источника высокомонохроматического излучения, чаще лазера, а первоначально – отдельных линий атомного спектра люминесцентной ртутной лампы низкого давления.
Спектр появляется в результате неупругих взаимодействий фотонов светового пучка с молекулами вещества. Фотон, сталкиваясь с электроном молекулы, может переводить его на более высокий молекулярный энергетический уровень, теряя при этом часть энергии. Появляющиеся линии называют стоксовыми. Возможен случай, когда фотон сталкивается с электроном, находящемся на высоком молекулярном энергетическом уровне, и переводит его на более низкую орбиталь, захватывая часть его энергии. Появляются линии, симметричные стоксовым относительно основной линии (падающий фотон) и называемые антистоксовыми. Стоксовские линии, т.е. менее энергичные, более интенсивны, т.к. процесс передачи энергии фотоном электрону более вероятен. Однако все линии КР- спектров малоинтенсивны по сравнению с возбуждающей (всего около 10-7 от общей интенсивности рассеянного света). Поэтому КР спектры фиксируют перпендикулярно направлению возбуждающего пучка. Регистрацию спектра осуществляют как обычно. При этом около основной возбуждающей линии n0 образуется ряд узких линий, соответствующих ni. По расстояниям между n0 и ni определяются величины Dn.
Вид спектра сходен с получаемым в ИК спектроскопии. В современных приборах возбуждение рассеянного света производится монохроматическим лазерным лучом, что позволяет при получении спектра обходиться 1÷10 мг вещества. Пробу можно вводить как в виде чистой жидкости или раствора, так и в виде твердого порошка.
Электромагнитные колебания возникают за счет движения зарядов. Соответственно, их поглощение связано со смещением зарядов. Очевидно, что прямое поглощение в ИК области будет происходить с достаточной интенсивностью, если связь полярна. В спектрах КР интенсивные полосы дают симметричные колебания неполярных связей, так как в этом случае важен дипольный момент, возникающий в процессе колебания. Следовательно, в простейших случаях в ИК спектрах должны проявляться колебания, неактивные в спектрах КР, и, соответственно, наоборот. Для симметричных молекул в ИК спектрах активны антифазные колебания, тогда как в спектрах КР - синфазные. По мере снижения симметрии молекулы многие колебания достаточно интенсивно проявляются в том и другом спектре. Следовательно, ИК- и КР- спектры дополняют друг друга, и при совместном применении этих методов может быть получена максимальная информация о частотах колебаний исследуемого вещества.
Полосы в колебательных спектрах делятся на два типа. Характеристические(в основном валентные) полосы, присутствие которых в спектре доказывает наличие в исследуемом веществе определенных структурных элементов.
Характеристическими являются те колебания, которые хотя бы по одному параметру (mr или F) существенно отличаются от основных колебаний С-С (это колебания легких атомов: С-Н, О-Н, N-Н или кратных связей).
Характеристическое колебание принадлежит определенной связи и, следовательно, имеет достаточно постоянную частоту в различных веществах, которая изменяется лишь незначительно за счет взаимодействия с остальной частью молекулы.
Нехарактеристическиеполосы, занимающие область 400÷1000 см-1, где проявляются многочисленные не поддающиеся отнесению валентные колебания связей С-С, С-N, N-О и деформационные колебания. Это область колебаний углеродного скелета молекулы, которая резко реагирует на малейшие изменения в структуре молекулы.Нехарактеристические колебания составляют основную часть спектра и для каждого вещества образуют свой, неповторимый набор полос. Нет двух соединений за исключением энантиомеров (оптических антиподов), которые имели бы одинаковые ИК спектры (и спектры КР). Этим часто пользуются для установления тождественности веществ, так как совпадение ИК спектров является убедительным доказательством идентичности исследуемых образцов.
Учитывая, что в спектре одного вещества всегда может быть найдена полоса, отсутствующая в спектре другого, возможен качественный анализ смесей, если спектры компонентов известны.
На том же основании может быть выполнен количественный анализ путем измерения интенсивностей соответствующих полос. Когда строение вещества уже установлено, в нехарактеристической области спектра некоторые полосы удается отнести к определенным колебаниям.
Однако перед исследователем стоит обычно противоположная задача - установить строение по спектру. В этом отношении возможности ИК спектроскопии не нужно переоценивать, следует использовать только абсолютно надежные критерии.
В частности, данные, полученные из рассмотрения области ниже 1500 см-1, нельзя расценивать как доказательства, а лишь как свидетельства в пользу присутствия того или другого структурного элемента. К ошибочным выводам может привести использование для структурных отнесений (в частности, для определения конформации и ближайшего окружения) малых изменений в величине характеристической частоты.
Иными словами, из колебательных спектров не следует пытаться извлечь информацию, достоверность которой сомнительна.
Для описания колебательных спектров чаще всего используют следующую информацию:
-колебания связи С-Н. Валентные колебания С-Н при насыщенном углеродном атоме проявляются в области 2800÷3000 см-1. Для ациклических и ненапряженных циклических структур nCH имеют следующие значения (в см-1):
| СН3 | 2962 см-1 |  2972 см-1 2972 см-1
|
| СН2 | 2853 см-1 |  2926 см-1 2926 см-1
|
| СН | 2890 см-1 |
Полосы характеристичны, но малоинформативны, так как в веществе обычно наблюдаются разные колебания С-Н, которые, кроме того, могут взаимодействовать между собой. Отдельные полосы колебаний накладываются друг на друга, образуя в области 2800÷3000 см-1 полосу, имеющую отдельные слабо выраженные максимумы. Для определения структуры вещества эти полосы могут оказаться полезными только в том случае, если в соединении мало атомов водорода, как, например, в полигалогеналканах. Отсутствие полос в данной области является убедительным доказательством отсутствия в веществе атомов водорода при насыщенных углеродных атомах.
Деформационные колебания dСН, расположенные в области 1350÷1470 см-1, малохарактеристичны, но обычно могут быть обнаружены в спектре:
| СН3 |  1375 см-1 1375 см-1
|  1450 см-1 1450 см-1
|
| СН2 | 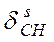 1475 см-1 1475 см-1
|
Достаточно характеристичным считается поглощение двух метильных групп при одном углеродном атоме (геминальное замещение), образующее два близких максимума (дублет) примерно равной интенсивности в области 1370÷1385 см-1.
Информативность спектров можно повысить используя различия в частотах колебаний связей, содержащих различные изотопные модификации атомов. В частности часто используют дейтерированные соединения, обогащенные дейтерием вместо протия.
При анализе соединений, меченных дейтерием, весьма характеристичной является полоса nCD 2100÷2160 см-1, расположенная в области, где практически отсутствуют другие полосы.
-колебания связи С=С. В соединениях с изолированной двойной связью vc=c находится при 1600- 1680 см-1.
В циклических системах, особенно в напряженных, значение этой частоты несколько ниже. Частота колебаний двойной связи заметно повышается с ростом степени ее замещенности, например:
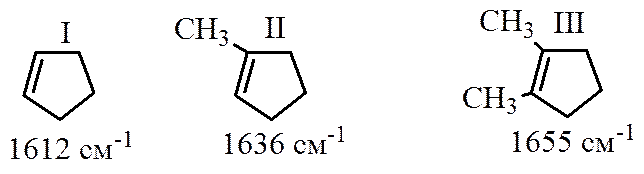
В ИК спектрах симметрично замещенных алкенов (неполярная двойная связь) nС=С проявляется полосой ничтожно малой интенсивности, как, например, в спектрах соединений (I) и (III); для несимметрично замещенной двойной связи (например, в соединении II) эта полоса достаточно интенсивна. В спектрах КР колебание С=С в любом случае активнее, чем в ИК спектре, и любая двойная связь дает мощную (обычно наиболее интенсивную в спектре) линию. О наличии в веществе двойной связи дополнительно может свидетельствовать характеристическая полоса (полосы) n=CH, расположенная в области 3000÷3100 см-1.
Деформационные колебания d=СН могут быть полезны для определения конфигурации заместителей при двойной связи: для цис- изомеров они расположены в области 650÷750 см-1, а для транс- изомеров - в области 960÷970 см-1.
Таким образом, на основании данных колебательных спектров (особенно спектра КР) может быть обнаружено присутствие в веществе изолированной двойной связи и сделаны определенные выводы о характере ее замещения.
Полоса n=СD весьма характеристична (2200÷2300 см-1) и позволяет уверенно отличить атом дейтерия, находящийся при двойной связи, от атома D при насыщенном углеродном атоме.
-колебания сопряженных диеновых систем.
Сопряженные диеновые системы в области 1500÷1650 см-1 имеют две полосы, соответствующие двум типам валентных колебаний - синфазному и антифазному, например: 
В целом полосы колебаний диеновых систем в ИК- и КР- спектрах значительно более интенсивны по сравнению с полосами изолированных двойных связей, особенно если диеновая система имеет трансоидную конфигурацию. В ИК спектре более активно колебание  , тогда как в спектре КР - колебание
, тогда как в спектре КР - колебание 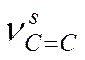 . В ИК спектре симметричных диенов (например, бутадиена) интенсивность полосы
. В ИК спектре симметричных диенов (например, бутадиена) интенсивность полосы  может быть исчезающе мала. При введении в диеновую систему алкильных заместителей значения частот
может быть исчезающе мала. При введении в диеновую систему алкильных заместителей значения частот 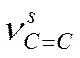 и
и  закономерно повышаются. Колебания n=CH в диенах проявляются в той же области, что и в алкенах (3000÷3100 см-1).
закономерно повышаются. Колебания n=CH в диенах проявляются в той же области, что и в алкенах (3000÷3100 см-1).
Таким образом, наличие в веществе диеновой системы относительно легко определяется по данным колебательных спектров. При сопряжении двойной связи с ароматическим ядром частота ее колебания смещается в низкочастотную область (на »30 см-1), при этом интенсивность поглощения повышается. При увеличении длины цепи сопряжения (в спектрах полиенов) растет общее число полос nС=С, причем частоты их колебаний уменьшаются, а интенсивность значительно возрастает.
-колебания ароматических систем. Валентные колебания С-С-связей бензольного ядра дают полосы умеренной интенсивности при 1585÷1600 см-1 и 1400÷1500 см-1, что делает их неудобными для идентификации, так как эта область близка к колебаниям nС=С. Колебания nCH аренов лежат в области 3020÷3100 см-1; обычно они проявляются в виде группы полос средней интенсивности, несколько большей, чем у поглощающих в той же области n=CH алкенов.
В спектрах ароматических соединений имеются интенсивные полосы неплоских деформационных колебаний С-Н в области 650÷900 см-1. Эта область дает некоторые возможности для определения числа и расположения заместителей в ароматическом ядре, а также взаимного расположения бензольных колец в полиядерных ароматических соединениях. Как правило, отсутствие сильных полос в области 650÷900 см-1 свидетельствует об отсутствии в веществе ароматического ядра. Кроме того, в этой области проявляются колебания связей углерод-галоген, причем полосы обычно имеют высокую интенсивность: C-Cl (550÷850 см-1), C-Br (515÷690 см-1), C-I (500÷600 см-1). Колебания связи C-F проявляются в области скелетных колебаний связей С-С, поэтому наблюдать их очень сложно. Использовать колебания связей углерод-галоген для определения галогенов в составе вещества не имеет смысла (есть множество методов более быстрых и точных), но для наблюдения промежуточных продуктов и взаимодействий в исследовании механизмов реакций появление полос может дать полезную информацию.
Для установления положения заместителей в ароматическом ядре иногда используют область 1650÷2000 см-1, где проявляются исключительно слабыми полосами обертоны и тоны более сложного происхождения. Полосы в этой области в зависимости от характера замещения имеют различный контур. Надежность данного признака невелика, и, кроме того, эта область полностью перекрывается при наличии в веществе карбонильной группы.
Колебательные спектры важнейших гетероциклических систем имеют много общего со спектрами производных бензола: так, для фурана, тиофена, пиррола и пиридина nCH 3010÷3080 см-1 и nC-С (кольцо) 1300÷1600 см-1, причем положение полосы vС-С существенно зависит от типа гетероцикла и характера замещения. В данной области может появиться от двух до четырех полос. Ниже приводятся основные частоты в спектрах важнейших гетероциклов (в см-1)
| фуран | 3125 - 3165 | 1500 - 1565 |
| тиофен | 3050 - 3125 | 1040 - 1520 |
| пиррол | 3400 - 3440 | 1500 - 1565 |
| пиридин | 3020 - 3070 | 1430 - 1650 |
-колебания связи СºС. Наличие связи обычно устанавливают по полосе валентных колебаний 2100÷2250 см-1, т.к. в этой области другие полосы практически отсутствуют. Полоса средней интенсивности, при симметричном замещении в ИК спектре она может стать практически незаметной, в КР спектре полоса активна всегда и ее интенсивность тем больше, чем менее симметричен алкин.
- колебания связи О-Н. В сильно разбавленных растворах, обеспечивающих отсутствие межмолекулярных взаимодействий, гидроксильные группы проявляются высокоинтенсивной полосой валентных колебаний 3200÷3600 см-1. Если гидроксогруппа участвует в водородной связи, то положение и характер полосы начинает сильно зависеть от степени вовлеченности, т.к. начинает меняться силовая постоянная связи. Если связь межмолекулярная, появляется широкая неструктурированная полоса перекрывающая весь диапазон 3200÷3600 см-1. Если наблюдается внутримолекулярная водородная связь, то об этом свидетельствует интенсивная полоса около 3500 см-1, смещенная в область низких частот по сравнению со свободными группами. Для избежания возможности образования межмолекулярных связей следует использовать малополярные растворители (углеводороды, CCl4) и концентрацию меньше 5×10-3 моль/л. Свободный фенольный гидроксил проявляется полосой валентного колебания 3600÷3615 см-1 высокой интенсивности.
Деформационные колебания гидроксогрупп расположены в области 1330÷1420 см-1 и малопригодны для идентификации. Димеры карбоновых кислот проявляются широкой интенсивной полосой в области 1200÷1400 см-1, но отнесение полосы может быть уверено сделано только после доказательства, что вещество действительно карбоновая кислота.
- колебания связи С-О. Связь проявляется в простых эфирах и спиртах интенсивной полосой области 1000÷1275 см-1. Сложные эфиры в спектрах содержат две полосы за счет группы С-О-С: симметричного колебания 1020÷1075 (более слабая в ИК спектре) и антисимметричного при 1200÷1275 см-1 (более слабая в КР спектре). В этом диапазоне проявляются полосы различных групп и полосы малохарактеристичны, но чаще всего именно они наиболее интенсивны.
- колебания связи С=О. Валентные колебания карбонильной группы присутствуют в спектрах различных соединений: альдегидов, кетонов, карбоновых кислот, ангидридов и т.д. Это всегда высокоактивный пик в области 1650÷1680 см-1, где прочие полосы практически отсутствуют. Это одна из наиболее характеристических полос, ее наличие или отсутствие может служить убедительным доводом наличия или отсутствия карбонильных групп. Конкретный диапазон проявления полосы зависит от соседних групп и группы в которую входит карбонил, индукционный эффект (-I) уменьшает длину связи С=О и, следовательно, увеличивается силовая постоянная и частота. Для альдегидов и кетонов полоса находится около 1710÷1750, карбоновых кислот - 1750÷1770 (мономеры) и 1706÷1720 (димеры), сложных эфиров - 1735÷1750, амидов кислот - 1650÷1695, хлорангидридов кислот - 1785÷1815, фторангидридов- 1865÷1875, ангидридов кислот - 1740÷1790 и 1800÷1850 см-1. Эффект сопряжения p-электронов понижает частоту колебания: в системах С=С-С=О и С6Н5-С=О полоса расположена около 1665÷1685 см-1.
Таким образом, спектры карбонильных соединений позволяют получить большой объем вполне однозначной информации, особенно учитывая прочие полосы: для сложных эфиров и ангидридов - полоса С-О, амидов - полоса N-H, в спектрах альдегидов часто присутствует полоса группы С(О)-Н около 2695÷2830 см-1. Для многих альдегидов и кетонов спектр представляет собой сумму основной и енольной формы.
Сводка спектральных проявлений различных группировок в ИК- и КР- спектрах приводена в таблице №2, однако существуют специальные таблицы, содержащие больший набор частот и позволяющие исследовать практические наборы полос от различных образцов.
Таблица №2 Основные частоты колебаний в ИК-спектроскопии
| Частота, см-1 | Интенсивность | Природа колебаний | Тип соединений |
| 3620- 3600 | с., ср. | nОН (своб.) | Разбавленные растворы спиртов |
| 3600- 3500 | с., ср. | nОН (связ.) | Внутримолекулярные водородные связи в спиртах |
| с., ср. | 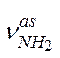 (своб.) (своб.)
| Разбавленные растворы первичных амидов | |
| 3400- 3350 | ср. | nNH(своб.) | Вторичные амины, N-замещенные амиды |
| 3550- 3520 | с., ср. | nOH(своб.) | Разбавленные растворы кислот |
| 3500- 3400 | с., ср. | nNH2(своб.) | Первичные амины, амиды |
| с. |  (своб.) (своб.)
| Разбавленные растворы амидов | |
| 3330- 3260 | ср. | nºCH | Однозамещенные алкины |
| 3300- 3280 | ср. | nNH(связ.) | N-однозамещенные амиды |
| 3200- 2500 | ср. | nОН(связ.) | Димеры кислот |
| 3100- 3020 | ср., сл. | nCH | Арены |
| 3100- 3000 | ср., сл. | n=CH | Алкены |
| с. | 
| Алканы | |
| 2930- 2910 | ср. | 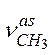
| СН3 при бензольном кольце |
| сл. | 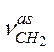
| Алканы | |
| сл. | nCН | Алканы | |
| 2880- 2860 | ср., сл. | 
| Алканы, СН3 при бензольном кольце |
| 2860- 2850 | ср. | 
| Алканы |
| 2695- 2830 | сл. | nC(O)Н | Альдегиды |
| 2250- 2100 | сл. | nСºС | Алкины |
| 2240- 2260 | ср. | nСºN | Нитрилы |
| 1850- 1650 | оч. с. | nС=О | Карбонильные соединения, кислоты и производные |
| 1680- 1600 | ср., сл. | nС=С | Алкены |
| 1600- 1585, 1500- 1400 | с., ср., сл. | nС-С | Арены |
| 1550- 1580 | ср., сл. | 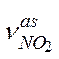
| Нитросоединения |
| ср. | 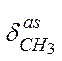
| Алканы | |
| 1450- 1300 | сл. | dСН | Замещенные алкены |
| 1420- 1330 | ср. | dОН | Спирты, фенолы, кислоты |
| 1385- 1370 | ср. | 
| гем- диметильная группа |
| 1385- 1375 | ср. | 
| Метилбензолы |
| 1380- 1370 | сл. | 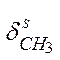
| Алканы |
| 1370- 1390 | с., ср. | 
| Нитросоединения |
| 1280- 1200 | с. | nСОС | Сложные эфиры |
| 1250- 1180 | ср. | nС-N | Третичные амины (ArNR2, (RCH2)3N) |
| 1220- 1125 | с. | nС-О | Вторичные, третичные спирты |
| 1200- 1160, 1145- 1105 | с., ср. | nС-О | Кетали, ацетали |
| 1150- 1050 | с. | 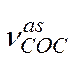
| Эфиры |
| 1085- 1050 | с., ср. | nС-О | Спирты |
| 970- 950 | ср. | dСН | Транс- алкены |
| 900-650 | с. | dСН | Арены |
| 750- 650 | ср. | d=СН | Цис- диены |
| Тип связи и соединений | Частота, см-1 | ||
| -С=С- | |||
| алкены | 1680- 1620 | ||
| цис- производные | 1665- 1635 | ||
| транс- производные | 1675- 1660 | ||
| циклические | 1650- 1550 | ||
| сопряженные | 1660- 1580 | ||
| -С=С=С- | |||
| аллены | 1970- 1940 (nas) | ||
| 1070- 1060 (ns) | |||
| -CºC- | |||
| Алкины | 2270- 2190 | ||
| -CºC-H | 2140- 2100 | ||
| ñС=О | |||
| Кетоны | алифатические | 1725- 1700 | |
| непредельные | 1690- 1660 | ||
| арилкетоны | 170- 1680 | ||
| диарилкетоны | 1670- 1660 | ||
| циклические | 1780- 1700 | ||
| Дикетоны | a | 1730- 1710 | |
| b | 1640- 1635 | ||
| Альдегиды | алифатические | 1740- 1720 | |
| непредельные | 1705- 1650 | ||
| ароматические | 1715- 1685 | ||
| Карбоновые кислоты | мономер | ||
| димер | 1725- 1700 | ||
| непредельные | 1715- 1680 | ||
| ароматические | 1700- 1680 | ||
| лактоны | 1850- 1720 | ||
| ангидриды | |||
| Сложные эфиры | алифатические | 1750- 1735 |
| непредельные | 1730- 1710 | |
| виниловые и ароматические | 1800- 1770 | |
| ñС=N | ||
| В открытой цепи | 1690- 1620 | |
| В цикле | 1660- 1480 | |
| a,b- непредельные | ||
| -CºN | ||
| Предельные нитрилы | 2265- 2240 | |
| a,b- непредельные | 2240- 2215 | |
| Арилнитрилы | ||
| Изонитрилы | 2185- 2120 | |
| -N=O | ||
| -O-N=O | 1680- 1610 | |
| -C-N=O | 1600- 1500 | |
| -N-N=O | 1500- 1440 | |
| -N=N- | ||
| Азосоединения | 1600- 1400 | |
| Азиды (-N3) | 2160- 2120 | |
| ñC=S | 1250- 1020 | |
| -S=O | 1070- 1010 | |
| -P=O | 1350- 1100 |
|
из
5.00
|
Обсуждение в статье: Колебательная спектроскопия |
|
Обсуждений еще не было, будьте первым... ↓↓↓ |

Почему 1285321 студент выбрали МегаОбучалку...
Система поиска информации
Мобильная версия сайта
Удобная навигация
Нет шокирующей рекламы