 |
Главная |


Общие методы получения фосфолипилов
|
из
5.00
|
Из растительного сырья
Сложность выделения фосфолипидов растения заключается в одновременном присутствии в нем ферментов-липаз, гидролизующих фосфолипиды, с образованием свободных жирных кислот. Поэтому выбор растворителя для экстракции фосфолипидов, одновременно ингибирующего их ферментатив-ное разложение имеет особое значение.
Наиболее часто для предварительной обработки раститель-ного материала используют изопропиловый спирт или ацетон.
В этом случае, воздушно-сухое сырье обрабатывают в течение 5-10 минут горячим изопропиловым спиртом. Горячую смесь фильтруют, остаток на фильтре промывают сначала горячим изопропиловым спиртом, затем смесью хлороформ - пропанол-2 (1:1) и хлороформом. Объединенные фильтраты упаривают под вакуумом, остаток растворяют в хлороформе. Полученный раствор несколько раз промывают водой очищенной и концентрируют.
Согласно альтернативному методу, фосфолипиды, из измельченных и обезжиренных ацетоном частей растения извлекают смесью хлороформ-метанол (2:1). Растворители отгоняют под вакуумом в мягких условиях, остаток растворяют в хлороформе и фильтруют; фильтрат сгущают до небольшого объема. Для предварительной очистки суммы фосфолипидов от сопутствующих веществ (масло, пигменты, углеводы, стероиды и др.) сухой экстракт обрабатывают ацетоном. Нерастворимый в ацетоне остаток растворяют в смеси хлороформ-метанол-вода (90:10:1) и для окончательной очистки от углеводов пропускают эту смесь через колонку с предварительно набухшим в той же смеси молселектом Г-25. Полноту очистки от углеводов проверяют методом ТСХ элюата в системе: хлороформ-метанол-вода 65:25:4.
Для фракционного разделения суммы фосфолипидов, полученный ранее экстракт растворяют в хлороформе и наносят на колонку с силикагелем. Элюирование компонентов осущест-вляют ацетоном, хлороформом, затем смесью хлороформ-метанол возрастающей полярности. При этом ацетоном извлека-ются вещества стероидного характера, хлороформом – нейтральные липиды. Смесь хлороформ-метанол (4:1) позволяет выделить фосфатидилэтаноламины и фосфатидилхолины; а тот же элюент состава (2:1) будет содержать фосфатидилхолины и фосфатидилинозиты, после чего в метанольную фракцию перейдут лишь лизофосфатидилхолины.
ХРОМОГЕННЫЙ КОМПЛЕКС
Около 10 г измельченного сырья (точная навеска) помеща-ют в колбу вместимостью 300 мл, прибавляют 200 мл воды очищенной и оставляют на 1 час при комнатной температуре. Затем колбу соединяют с обратным холодильником, кипятят в течение 2 ч, охлаждают, фильтруют в мерную колбу вмести-мостью 300 мл, сырье переносят на фильтр и промывают теплой водой. Водное извлечение в колбе охлаждают до температуры 18-200С, доводят объем извлечения водой очищенной до метки и тщательно перемешивают.
25 мл фильтрата переносят во взвешенную фарфоровую чашку, выпаривают на водяной бане досуха и сушат при темпе-ратуре 100-1050С в течение 3 ч, затем охлаждают в эксикаторе и быстро взвешивают, определяя массу сухого остатка.
Для определения хромогенного комплекса 100 мл фильтрата помещают в стакан вместимостью 150 мл, подкисляют 25% раствором кислоты хлороводородной (0.5-0.8 мл) до рН=1.0-2.0 по универсальной индикаторной бумаге, перемешивают и оставляют на 30 мин. После выпадения темно-бурого осадка содержимое стакана фильтруют через бумажный фильтр.
25 мл фильтрата, полученного после осаждения хромогенного комплекса кислотой хлороводородной, переносят в высушенную до постоянной массы и взвешенную фарфоровую чашку, выпаривают на водяной бане досуха и сушат при температуре 100-1050С в течение 3 ч, затем охлаждают в эксикаторе и быстро взвешивают, определяя массу сухого остатка без хромогенного комплекса.
Содержание хромогенного комплекса в процентах (Х) в пересчете на абсолютно сухое сырье, вычисляют по формуле:
|
m . 25 . (100 - W)
где m1 - масса сухого остатка до осаждения кислотой хлороводородной, в граммах;
m2 - масса сухого остатка после осаждения кислотой хлороводородной, в граммах;
m - масса навески сырья, в граммах;
W - потеря в массе при высушивании сырья, в процентах [7].
ЭФИРНЫЕ МАСЛА
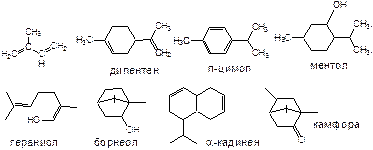 |
Эфирные масла – смесь душистых летучих веществ растений различных групп: производные углеводородов, спирты, кетоны, альдегиды, лактоны, кислоты, фенолы, простые и сложные эфиры и т.д.
Наиболее богаты эфирными маслами растения семейств: астровые, сельдерейные, розоцветные, лавровые, яснотковые, цитрусовые.
Эфирные масла практически не растворимы в воде, растворимы в жирных и минеральных маслах, спирте, эфире, других органических растворителях. Температуры их кипения колеблются от 140 до 2600С, они оптически активны.
Выделение:
2-3 г свежих измельченных эфиросодержащих частей растения (цветки, плоды) заливают 30 мл эфира петролейного (40-700С), и настаивают в течение суток, фильтруют.
Для проведения качественного анализа используют 2-3 мл раствора.
Качественный анализ [7,10-12]:
q Добавляют 1 каплю раствора пара-диметиламинобенз-альдегида (1 г пара-диметиламинобензальдегида смачивают 4 каплями воды, затем добавляют 3 мл кислоты серной концентрированной), появляется яркое окрашивание (терпены различного типа, гликозиды), желтое окрашивание, переходящее в малиново-красное от добавления 2-3 капель воды (ментол).
q
| |
q Добавляют 1-2 капли 1% раствора ванилина в кислоте серной концентрированной, появляется красно-фиолетовое или пурпурно-красное окрашивание (терпены с С3-ОН, C3-О-caxap). Желтое окрашивание, переходящее в малиново-красное от добавления 1 мл воды (ментол).
q Гидроксамовая проба. Добавляют 1% спиртовый раствор гидроксиламина солянокислого и раствора калия едкого до рН=8, затем несколько капель кислоты хлороводородной,1-2 капли 1% спиртового раствора железа окисного хлорида, появляется фиолетовое окрашивание (терпены, сложные эфиры).
q Реакция Сабетая. Добавляют 1 мл 1% раствора брома в хлороформе, появляется окрашивание от голубого до синего (сесквитерпены).
Хроматографический анализ:
Методами хроматографии в настоящее время определяют не только полный состав компонентов эфирных масел, но и доминирующий компонент масла (в эвкалипте – цинеол, в мяте – ментол, в фенхеле – фенхон, в кориандре – линалол и т.д.).
Наиболее подходящими системами для ТСХ хроматографии эфирных масел являются:
1) петролейный эфир (40-700С)
2) хлороформ – метиловый спирт от (4:1) до (19:1);
3) хлороформ – этиловый спирт (20:1);
4) хлороформ - ацетон (9:1);
5) толуол – этилацетат (1:1);
6) н-гексан - этилацетат (1:1).
Поскольку эфирные масла не однородны по составу, то оптимальным методом оценки качественного и количественного состава является метод газовой или газо-жидкостной хроматографии.
Для проведения газохроматографического исследования компонентов эфирных масел в хроматограф испаряют при температуре 200-2500С пробу эфирного масла. Под действием постоянно протекающего через эту трубку газа-носителя (обычно гелий или водород) эфирное масло в виде пара движется по колонке с SE-30, Carbowax 20M, Chrompack CP Sil 5CB или Chrompack CP-Wax 52CB сорбентом. Температура колонки в ходе анализа повышается от 600С до 2100С со скоростью 3-4 градуса/мин. Выход компонентов масла обычно регистрируется пламенно-ионизационным детектором [142,143]
Для идентификации эфирных масел используют следующие показатели:
- цвет, прозрачность, вкус;
- температура застывания и кипения;
- содержание жирных и минеральных масел;
- плотность;
- угол вращения;
- показатель преломления;
- кислотное число;
- эфирное число;
- содержание сложных эфиров, фенолов, спиртов.
Количественное определение:
Эфирные масла, в зависимости от их химической природы и содержания, определяют различными методами.
Метод 1. Около 1 г (точная навеска) измельченного сырья помещают в круглодонную колбу вместимостью 300 мл, приливают 100 мл горячей воды, присоединяют обратный холодильник с градуированной насадкой и нагревают 2-4 ч, в зависимости от содержания эфирных масел в сырье, которые накапливаются по мере их отгонки с паром в градуированной пробирке.
Объем масла замеряют после охлаждения прибора до комнатной температуры.
Содержание эфирного масла в объемно-весовых процентах (Х) вычисляют по формуле:
|
m . (100 - W)
где V – объем эфирного масла, в миллилитрах;
m – масса навески сырья, в граммах;
W – потеря в массе при высушивании сырья в процентах.
* Для извлечения эфирных масел из сырья предложено использовать ксилен, пентан, гептан, хлороформ, четырех-хлористый углерод [22].
Если масло в процессе отгонки загустевает или образует эмульсию, более пригодным является прибор с внешней градуированной пробиркой.
Перед каждым определением через прибор пропускают пар в течение 15-20 мин. После 6-8 определений прибор необходимо промыть последовательно ацетоном и водой очищенной [7,93,144].
Метод 2.Точную навеску препарата помещают в мерную колбу вместимостью 10 мл, прибавляют 1 мл раствора 1,8-цинеола (внутренний стандарт), доводят объем раствора спиртом метиловым до метки, перемешивают и фильтруют через фильтр с размером пор 0.45 мкм (испытуемый раствор).
По 2 мкл испытуемого раствора, раствора СО фенхона, линалола или др. растворов (жидкого экстракта фенхеля, жидкого экстракта кориандра) попеременно хроматографируют на газовом хроматографе с пламенно-ионизационным детектором, получая не менее 3 хроматограмм для каждого из растворов в следующих условиях:
§ колонка кварцевая капиллярная DB-WAXETR [30 х 0.53];
§ газ-носитель - аргон;
§ толщина неподвижной фазы – 1.00 мкм;
§ скорость газа-носителя – 30 мл/мин;
§ начальная температура колонки – 700С (10 мин);
§ скорость линейного нагрева колонки 60С/мин;
§ промежуточная температура колонки – 1700С (0 мин);
§ скорость линейного нагрева колонки – 30С/мин;
§ конечная температура колонки – 2000С (20 мин);
§ температура испарителя – 2200С, детектора – 2400С;
Содержание эфирного масла (Х) в 100 г препарата, в граммах, вычисляют по формуле:
|
S1 . S4 . 10 . m
где S1 – среднее значение площадей пиков 1,8-цинеола, вычисленное из хроматограмм испытуемого раствора;
S2 – среднее значение площадей пика фенхона (линалола) или др. СО, вычисленное из хроматограмм испытуемого раствора;
S3 – среднее значение площадей пика 1,8-цинеола, вычисленное из хроматограмм раствора препарата (жидкого экстракта фенхеля/кориандра или др.);
S4 – среднее значение площадей пика фенхона (линалола) или др. СО, вычисленное из хроматограмм раствора препарата (жидкого экстракта фенхеля/кориандра или др.);
m0 – масса навески жидкого экстракта фенхеля (кориандра), в граммах;
m – масса навески препарата, в граммах.
Примечания. Приготовление раствора 1,8-цинеола (внутренний стандарт). 80.1 мг 1,8-цинеола помещают в мерную колбу вместимостью 25 мл, доводят объем раствора спиртом метиловым до метки и перемешивают.
Приготовление раствора СО фенхона (линалола). 162.3 мг СО фенхона и 230.9 мг СО линалола помещают в мерную колбу вместимостью 50 мл, доводят объем раствора спиртом метиловым до метки, перемешивают (раствор 1). 1.0 мл раствора 1 помещают в мерную колбу вместимостью 100 мл, доводят объем раствора спиртом метиловым до метки, перемешивают (раствор 2). 2.0 мл раствора 2 помещают в мерную колбу вместимостью 10 мл, прибавляют 1 мл раствора 1,8-цинеола (внутренний стандарт), доводят объем раствора спиртом метиловым до метки, перемешивают.
Приготовление раствора жидкого экстракта фенхеля. 1.9584 г (точная навеска) СО жидкого экстракта фенхеля помещают в мерную колбу вместимостью 10 мл, прибавляют 1 мл раствора 1,8-цинеола (внутренний стандарт), доводят объем раствора спиртом метиловым до метки, перемешивают и фильтруют через фильтр с размером пор 0.45 мкм.
Приготовление раствора жидкого экстракта кориандра. 1.8925 г СО жидкого экстракта кориандра помещают в мерную колбу вместимостью 10 мл, прибавляют 1 мл раствора 1,8-цинеола (внутренний стандарт), доводят объем раствора спиртом метиловым до метки, перемешивают и фильтруют через фильтр с размером пор 0.45 мкм.
Время удерживания: 1,8-цинеола – 8.864 мин, фенхона – 16.798 мин, линалола – 20.708 мин.
Содержание доминирующего компонента в масле определя-ют по методу 3. В колбу вместимостью 100 мл с шейкой, градуированной на 10 мл с точностью до 0.1 мл, вносят 3 мл испытуемого препарата и 75 мл раствора резорцина. Смесь взбалтывают в течение 15 мин и после отстаивания доливают такое количество раствора резорцина, чтобы оставшееся масло собралось в градуированную часть колбы. Спустя 1 час отсчитывают объем непрореагировавшего масла. Отмеривание масла для анализа и отсчет непрореагировавшего масла производят при одинаковой температуре.
Содержание доминирующего компонента в масле в объемных процентах (Х) вычисляют по формуле:
|
где а – количество непрореагировавшего масла, в миллилитрах [145].
Содержание эфирных масел в препарате «Настойка мяты перечной» определяют аналогично описанной выше методике, но вместо резорцина используют смесь натрия хлорида и кислоты серной.
10 мл препарата помещают в колбу для определения эфирного масла вместимостью 100 мл, прибавляют 10 мл кислоты серной разведенной и 60 мл насыщенного раствора натрия хлорида. Колбу закрывают пробкой, энергично встряхивают в течение 5 мин и дают отстояться в течение 1-2 часов при 25-300С. Когда все эфирное масло выделится на поверхности жидкости, осторожно добавляют насыщенный раствор натрия хлорида до тех пор, пока слой эфирного масла не окажется в пределах градуированной части колбы, дают отстояться до полного отделения масла от водного слоя [13,146].
|
из
5.00
|
Обсуждение в статье: Общие методы получения фосфолипилов |
|
Обсуждений еще не было, будьте первым... ↓↓↓ |

Почему 1285321 студент выбрали МегаОбучалку...
Система поиска информации
Мобильная версия сайта
Удобная навигация
Нет шокирующей рекламы