 |
Главная |


ПРИОНЫ НАРУШАЮТ ЦЕНТРАЛЬНУЮ ДОГМУ?
|
из
5.00
|
На фоне этих представлений вполне еретически прозвучало открытие инфекционных белков-прионов, являющихся переносчиками ряда нейродегенеративных заболеваний человека и животных [6]. Основной вклад в концепцию инфекционных белков-прионов связывают с именем С. Прусинера, удостоенного за это Нобелевской премии в 1997 г. [7].
Из всех этих неизлечимых смертельных заболеваний человека и животных наиболее известно в последнее время так называемое коровье бешенство, оно же - губчатая болезнь мозга коров, или BSE (Bovine SponGIForm Encephalopathy).
Куру - первое, наиболее подробно исследованное в конце 50-х годов Д.К. Гайдушеком заболевание этого типа у человека, распространившееся среди членов племени Форе (Новая Гвинея) в результате ритуального каннибализма.
Кроме того, существуют еще три синдрома: болезнь Кройцфельда-Якоба, болезнь Герштона-Штресслера-Шейнкера и смертельная семейная бессонница.
РгРC РгРSc
Аналогичные болезни известны у целого ряда млекопитающих, прежде всего у овец и коз (а также у мышей, хомяков, кошек и др.) под названием скрэпи, или почесуха. Возбудителем всех этих заболеваний является инфекционный белок-прион, обозначаемый как РгРSc (от Scrapi). Этот белок имеет нормальный клеточный гомолог, то есть неболезнетворный и неинфекционный белок, обозначаемый как РгРC (от Cellular, то есть клеточный) [8]. Слово прион возникло как модифицированное сокращение английского: Protenacious Infection - белковая инфекция.
Особую "популярность" эти заболевания приобрели в связи с данными об инфекционности коровьего бешенства для человека. В последние годы идентифицирована новая разновидность болезни Кройцфельда-Якоба у человека, идентичная по некоторым молекулярным характеристикам BSE [9, 10]. Это имеет печальный эпидемиологический аспект, а также акцентирует возможность межвидового переноса прионной инфекции, что в общем-то было известно уже с начала 80-х годов из экспериментов с животными, прежде всего с мышками и хомячками, если не считать, что еще раньше Гайдушек показал заразность куру для обезьян [6, 11].
Характерным проявлением всех этих заболеваний на последних стадиях их развития служит образование в тканях головного и спинного мозга миелоидных тяжей и бляшек, состоящих из упомянутого прионного белка РгРSc. При этом ткани мозга на срезе приобретают вид губки. Отсюда и наименование "губчатая болезнь мозга".
Любая из перечисленных болезней может иметь три варианта возникновения:
а) инфекционный (уже упомянутый),
б) наследственный,
в) спорадический, то есть появление, казалось бы, без видимых причин, когда не просматривается ни наследственной предрасположенности, ни инфекции.
МОЛЕКУЛЯРНАЯ ГЕНЕТИКА ПРИОНОВ МЛЕКОПИТАЮЩИХ
Инфекционное начало оказалось ассоциированным именно с упомянутыми белковыми образованиями головного или спинного мозга. При этом белок-прион (РгРSc) из этих высокомолекулярных агрегатов и растворимый неинфекционный белок (РгРC) здоровых особей имеют одинаковую первичную структуру, то есть одинаковую аминокислотную последовательность, но различаются по вторичной и третичной структуре, то есть характером укладки полипептидной цепи (рис. 2).

Рис. 2. Предполагаемое изменение характера укладки полипептидной цепи
при превращении белка РгРC (а) в прион РгРSc (б)
В белке РгРSc обнаруживается существенно больше так называемых b-слоев - около 43% (в РгРC -3%)-и несколько уменьшается количество (43-34%) так называемых a-спиралей. При этом инфекционный белок, образующий уже упомянутые амилоидные тяжи, оказывается более кислотоустойчивым, термоустойчивым и более устойчивым к протеолизу, нежели его клеточный гомолог РгРC. Оказалось, что инфекционный белок возникает путем модификации своей структуры из обычного клеточного белка и при этом приобретает свойство инфекционности [8].
На короткий период конца 70-х - начала 80-х годов возникла гипотеза белковой наследственности, то есть наследственности без ДНК, коль скоро инфекционный агент - белок, а он к тому же еще и накапливается в зараженном организме, то есть воспроизводится. Эта сенсация просуществовала недолго, так как вскоре был открыт ген, который кодирует первичную структуру, то есть аминокислотную последовательность белка-приона и его клеточного предшественника. Ген оказался очень консервативным, то есть очень похожим у всех млекопитающих [12]. Функция его до сих пор точно неизвестна. Зато известно, что он необходим для развития заболевания. Мыши, лишенные этого гена, вполне жизнеспособны и практически здоровы [13], но не заболевают, если их инфицировать белком-прионом [14].
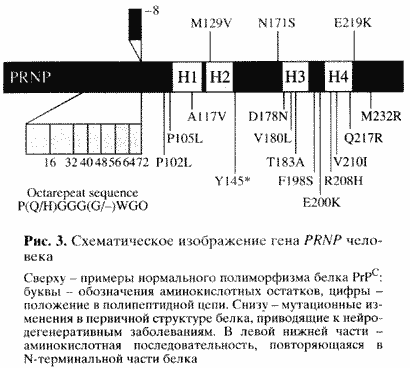
| На рис. 3 показана структура этого гена (PRNP) человека. Сверху - известные варианты нормального полиморфизма, то есть аминокислотные замены, не приводящие к заболеванию. Внизу - мутационные замены, приводящие к нейродегенеративным заболеваниям [8]. Особый интерес представляет начальный конец гена и соответственно так называемый N-терминальный конец белка. Здесь закодированы пять повторов из восьми аминокислот. Уменьшение числа повторов на один не сопровождается патологическими последствиями, в то время как увеличение числа повторов (известны варианты вплоть до 12 повторов) приводит к одной из наследственных форм заболевания [8]. Следовательно, эти повторы играют существенную роль в организации структуры белка и каким-то образом влияют на возможность его превращения в инфекционную форму. |
Итак, наследственные варианты заболевания - результат мутационного изменения первичной структуры белка, облегчающего превращение его в прион и далее - превращение всех вновь синтезируемых полипептидов в прионную, патогенную и инфекционную форму.
Спорадическая форма заболевания - спонтанное изменение характера укладки полипептидной цепи, превращающее клеточный белок в прион, а далее - опять же превращение всех вновь синтезируемых полипептидов в прионную, то есть инфекционную и патогенную, форму. Здесь очень важно отметить, что при одной и той же первичной структуре, то есть одной и той же аминокислотной последовательности, без всяких изменений кодирующего гена может возникнуть несколько вариантов укладки приона, и эти различающиеся варианты укладки воспроизводятся при последующей инфекции. Они получили название штаммов или правильнее - клонов приона [15].
И, наконец, инфекционная форма прионных заболеваний - результат проникновения в организм, в клетку млекопитающих инфекционного белка, возникшего любым из двух упомянутых способов.
В стандартные концепции легко укладывается возникновение наследственных форм заболевания. Можно представить, что одна первичная структура служит основой для разных способов пространственной укладки белковой молекулы. Чего мы не понимаем - как одна форма укладки сменяет другую и как далее эта укладка воспроизводится. То, что это явление (воспроизведение пространственной структуры, или конформации) имеет место, доказывают изящные эксперименты с так называемыми трансгенными мышами, у которых собственный ген PRNP заменен на гомологичный ген человека (рис. 4).

Можно взять прион от людей, погибших от разных вариантов прионных заболеваний. Такие белки различаются по физико-химическим свойствам, выявляемым в электрофорезе, то есть по подвижности в электрическом поле после их частичного разрушения при помощи протеолиза [16, 17]. Если этими различающимися формами белка человека заразить такую мышь, то она рано или поздно заболеет (инкубационный период - около 150 дней) и можно выделить белок-прион с исходными характеристиками инфекционного начала [18]. Таким образом, в результате модификации пространственной структуры белка могут появляться штаммы, или клоны, прионов. Механизм этого явления остается загадкой.
|
из
5.00
|
Обсуждение в статье: ПРИОНЫ НАРУШАЮТ ЦЕНТРАЛЬНУЮ ДОГМУ? |
|
Обсуждений еще не было, будьте первым... ↓↓↓ |

Почему 1285321 студент выбрали МегаОбучалку...
Система поиска информации
Мобильная версия сайта
Удобная навигация
Нет шокирующей рекламы