 |
Главная |


Частота некоторых генных болезней
|
из
5.00
|
| Заболевание | Частота |
| Синдром Мартина-Белл | 0,5 : 1000 |
| Муковисцидоз | 1 : 2000 |
| Миодистрофия Дюшенна | 1 : 3000-3500 ( у мужчин) |
| Синдром Марфана | 1 : 5000 |
| Адреногенитальный синдром | 1 : 10000 |
| Нейрофиброматоз Реклингаузена | 1 : 3000-5000 |
| Синдром Элерса-Данло | 1 : 5000 |
| Фенилкетонурия | 1 : 10000 |
| Гемофилия А | 1 : 10000 |
| Недостаточность a-антитрипсина | 1 : 3000-20000 |
| Синдром Луи-Барр | 1 : 30000-50000 |
| Галактоземия | 1 : 35000-50000 |
| Альбинизм | 1 : 40000 |
| Недостаточность глюкозо-6-фосфат ДГ | 1 : 4-20 ( у мужчин некоторых популяций) |
| Алкаптонурия | 1 : 100000 |
| Гомоцистинурия | 1 : 150000 |
больного (синдром Хартнупа), или по основному симптому (альбинизм, синдром ²голубых пеленок).
Когда научились регистрировать основные звенья патогенеза, то появились следующие термины: если обнаруживалось увеличение концентрации ключевого вещества в крови, то к его названию добавляли –емия (галактоземия), если в моче – урия (фруктозурия, фенилкетонурия).
Если основным биохимическим признаком служит накопление соединения в клетках, то в его термине появляется суффикс – оз (тирозиноз, холестериноз, ганглиозидоз). В последние годы стали указывать точное место повреждения – недостаточность аденозиндезаминазы, непереносимость дисахаридов, недостаточность глюкозо-6-фосфат ДГ.
Как известно из менделевской генетики, разные аллели могут проявляться в следующих вариантах: доминантном, рецессивном, кодоминантном. Чаще регистрируются первых два вида. Отсюда, в зависимости от генетического принципа классификации (типа наследования) выделяют несколько групп моногенных болезней.
Аутосомно-доминантные – дефектный ген расположен в одной из 22 пар аутосом, для недуга достаточно унаследовать мутантный аллель одного из родителей (нейрофиброматоз Реклингаузена, гемохроматоз, ахондроплазия, семейная гиперхолестеринемия, синдромы Марфана, Элерса-Данло и др.). Для большинства подобных повреждений характерна такая клиника, которая не наносит серьезного ущерба здоровью человека и часто не влияет на его способность иметь потомство (рис. 4).
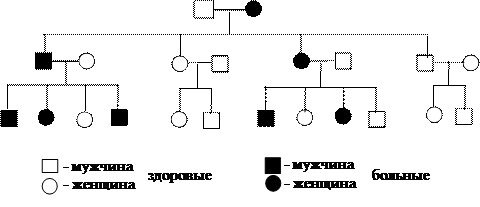 |
Рис. 4. Родословная с аутосомно-доминантным типом наследования болезни
(Синдром Марфана).
Характерными признаками данного типа являются:
- заболевание передается из поколения в поколение регулярно без пропусков (в родословной прослеживается по вертикали);
- риск рождения больного ребенка (при наличии патологии у одного из родителей) составляет 50%;
- здоровые индивиды имеют здоровых потомков;
- у больного ребенка обязательно болен хотя бы один из родителей;
- от двух больных родителей могут рождаться гомозиготы; болезнь у таких детей протекает тяжелее, чем у гетерозигот;
- оба пола поражаются с одинаковой частотой.
Аутосомно-рецессивные проявляются только у гомозигот. Известно более 1600 подобных болезней (галактоземия, муковисцидоз, болезнь Тея-Сакса, фенилкетонурия, адреногенитальный синдром и др.).
Наиболее типичные признаки (рис. 5):
- родители больного здоровы, но аналогичное заболевание регистрируется у родственников (передается в родословной по горизонтали) (в одном поколении);
- если больны оба родителя, то все дети будут поражены;
- у больного родителя рождаются здоровые дети;
- мальчики и девочки заболевают одинаково часто;
- риск рождения больного ребенка составляет 25% (соотношение больных и здоровых 1:4);
- кровнородственные браки увеличивают вероятность рождения больных в семье.

Рис. 5. Родословная с аутосомно-рецессивным типом наследования болезни
(синдром Тея-Сакса).
Х-сцепленные рецессивные – в своей основе имеют дефект гена, локализованного в половой Х-хромосоме (синдром Леша-Найхана, недостаточность глюкозо-6-фосфатДГ, синдром Барта). Женщина, унаследовав от одного из родителей патологический аллель, является гетерозиготой, т.е. фенотипически здорова; страдают только мужчины.
Ключевые компоненты данного типа (рис. 6):
- заболевание развивается у мужчин по материнской линии;
- у больных мальчиков могут быть больные братья и дяди по матери;
- сыновья никогда не наследуют недуг отца;
- у больного отца все дочери здоровы, но являются гетерозиготными
носителями мутантного гена;
- если женщина – гетерозиготный носитель, то половина ее сыновей больны, все дочери здоровы, причем половина из них гетерозиготы.

Х-сцепленные доминантные обусловлены тем, что у женщин две Х-хромосомы, а у мужчин одна. Женщина, унаследовав от одного из родителей патологический аллель, является гетерозиготой, а мужчина - гомозигота.
 Рис. 7. Родословная с Х-сцепленным доминантным типом наследования болезни (витамин Д-резистентный рахит). Обозначения те же, что и к рис.4.
Рис. 7. Родословная с Х-сцепленным доминантным типом наследования болезни (витамин Д-резистентный рахит). Обозначения те же, что и к рис.4.
Признаками, характерными для подобной патологии, являются (рис. 7):
- поражаются и мужчины, и женщины, но последних два раза больше;
- у больного обязательно страдает один из родителей;
- у больного отца все дочери больны, а сыновья здоровы;
- у больной матери равновероятно рождение больных и дочерей, и сыновей;
- у здоровых родителей все дети здоровы;
- в среднем женщины болеют менее тяжело, чем мужчины.
У-сцепленный тип наследования. Длительное время полагали, что У-хромосома содержит только гетерохроматиновые участки (без генов). Новейшие исследования позволили обнаружить и локализовать в этой структуре ряд генов, отвечающих за сперматогенез, контролирующих рост тела, конечностей, зубов, определяющих оволосение ушной раковины. Дефектный признак передается всем мальчикам. Естественно, все патологические мутации, затрагивающие сперматогенез наследоваться не могут, т.к. индивиды стерильны.
Среди описанных мутантных генов на аутосомно-доминантный характер наследования приходится 2/3, около 25% - на аутосомно-рецессивный, 6% дефектных транскриптонов локализованы в Х-хромосоме, 0,8% в У-хромосоме.
Выявлено 59 поврежденных генов в митохондриальной ДНК (митохондриальный тип наследования) (гл. 2).
Клинический принцип классификации генных болезней основывается на поражении той или иной системы либо органа. Согласно его положениям выделяют генные болезни соединительной ткани (мукополисахаридозы, болезнь Марфана и др.), ЖКТ (непереносимость дисахаридов, гиповитаминоз В12 из-за нарушения всасывания витамина и др.), почек (витамин Д-резистентный рахит, цистинурия и др.), печени (синдромы Криглера-Найяра, Жильбера и др.) или клеток крови: эритроцитов (недостаточность глюкозо-6-фосфатДГ), лейкоцитов (недостаточность аденозиндезаминазы). Сюда же можно отнести тезаурисмозы (болезни накопления различных веществ в органоидах клеток).
В связи с нарушением активности лизосомальных ферментов (лизосомные болезни) в клетках или межклеточном веществе накапливаются недеградированные биополимеры: гликозаминогликаны (мукополисахари-дозы); а если подобное повреждение локализуется в гепатоцитах или миоцитах, где подавляется активность энзимов, разрушающих гликоген, развиваются гликогенозы.
Другие внутриклеточные структуры – пероксисомы, главная функция которых деградация пероксидов, – также могут являться точкой приложения первичного действия мутантного гена. В этих случаях возникают так называемые пероксисомные болезни. Описано около 30 нозологических форм.
Однако более часто используется классификация, учитывающая характер повреждений первичного звена в метаболизме. По такому принципу различают:
-болезни обмена аминокислот (алкаптонурия, альбинизм, гистидинемия, фенилкетонурия и др.);
-болезни обмена углеводов (галактоземия, гликогенозы, фруктоземия и др.);
-болезни обмена липидов (ганглиозидозы, цереброзидозы, сульфатидозы, наследственная гиперхолестеринемия и др.);
-болезни обмена пуринов и пиримидинов (синдром Леша-Найхана, недостаточность аденозиндезаминазы, оротацидурия и др.);
-болезни обмена порфиринов (порфирии);
-болезни обмена билирубина (синдромы Криглера-Найяра, Ротора, Жильбера и др.);
-болезни метаболизма витаминов (витамин Д-резистентный рахит, пеллагрические поражения при дефиците витамина В6 и др.);
-болезни минерального обмена (синдром Менкеса, болезнь Вильсона-Коновалова и др.);
-болезни компонентов крови: форменных элементов (гемоглобинозы, талассемии и др.) или белков плазмы (анальбуминемия).
В последние годы среди причин наследственных болезней стали указывать на отсутствие восстановления нарушенных участков в структуре гена. Как известно, мицеллы ДНК обладают способностью к устранению повреждений нуклеотидной последовательности (репарации). С помощью нуклеаз измененные фрагменты вычленяются (рестрикция), затем соответствующая полимераза достраивает участок цепи, а ДНК-лигаза связывает его с основной нитью. Но появились факты, свидетельствующие о том, что подобные явления в клетках человека могут быть причиной возникновения ряда заболеваний, приводящих к тяжелой инвалидности или гибели. Возник даже термин «болезни репарации». Среди них выделяют три группы:
1) пигментная ксеродермия; у носителей этой патологии под действием обычного солнечного света возникают сначала красные пятна, которые постепенно переходят в незарастающие коросты, часто трансформирующиеся в раковые опухоли. В 1968 году Д. Кливер нашел причину этой коварной болезни – дефекты разных репарирующих систем;
2) синдром ломкости хромосом, или синдромы спонтанной хромосомной нестабильности:
а) синдром Блума, характеризуется светобоязнью, повышенной чувствительностью кожи к УФО;
б) синдром Луи-Барр (гл. 8.4) проявляется нарушением походки, телеангиоэктазиями (локальным и чрезмерным расширением капилляров и мелких сосудов);
в) анемия Фанкони(anaemia Fanconi), наследуется по аутосомно-рецессивному типу, симптомы включают низкий рост, микроцефалию, косоглазие, деформацию скелета, глухоту, панцитопению, лейкопению, эритроцитопению, также признаки малокровия: одышку, слабость, бледность, тахикардию (гл.7);
3) болезни преждевременного старения (прогерии): а) синдром Вернера (progeria adultorum, morbus Werner) начинается в подростковом возрасте эндокринными нарушениями (сахарным диабетом, гипогонадизмом), присоединяются трофические язвы, склеродермия, для больных характерны маленькие стопы, кисти, старческий вид; б) синдром Коккейна (syndrome Cockayne) наследуется по аутосомно-рецессивному типу; к 2005 году диагностировано 180 случаев. Причина - дефект гена, кодирующего белок, который участвует в процессе репарации ДНК. Первые симптомы появляются на 2-3-м годах жизни в виде повышения фоточувствительности кожи, позднее присоединяются отставание в росте (карликовость), в массе тела (вплоть до кахексии), нарушения походки, возникают контрактуры в суставах, не пропорционально увеличиваются уши, конечности, стопы, кисти, снижается интеллект, развивается преждевременное старение. Смерть наступает в 20-30 лет от осложнений атеросклероза (гиперхолестеринемии, накопления ЛПНП).
К прогериям принадлежит и еще одна очень редкая наследственная аномалия сенильный нанизм, известный с 1886 года как синдром Хатчинсона-Гилфорда ((syndrome Hutchinson-Gilford). Тип наследования аутосомно-рецессивный. У детей – старческая внешность, редкие седые волосы, тонкая морщинистая кожа, карликовый рост, часто - гипогонадизм, остеопороз, слабость скелетной мускулатуры, но IQ в пределах нормы. Локализация генетического дефекта не установлена. Продолжительность жизни не превышает 20 лет.
В диагностике подобных заболеваний выясняют способность лимфоцитов к репарации или их чувствительность к действию физических мутагенов (УФО, g-лучей). Полученные результаты приобретают выраженную актуальность для таких больных, т.к. для них особенно опасны экологически вредные воздействия, неадекватное питание с дефицитом витаминов А, Е, С, обусловливающим недостаточность антиоксидантной защиты. Все это необходимо учитывать при проведении диагностических и лечебных мероприятий (жесткое дозирование рентгеновского облучения, физио- и химиотерапии) лицам, страдающим болезнями репарации.
|
из
5.00
|
Обсуждение в статье: Частота некоторых генных болезней |
|
Обсуждений еще не было, будьте первым... ↓↓↓ |

Почему 1285321 студент выбрали МегаОбучалку...
Система поиска информации
Мобильная версия сайта
Удобная навигация
Нет шокирующей рекламы