 |
Главная |


Определение энтальпии нейтрализации
|
из
5.00
|
Студент получает у преподавателя один из следующих вариантов задания (см. табл.2).
Реакционный сосуд после определения постоянной калориметра несколько раз ополаскивают дистиллированной водой и пипеткой вносят в него 25 см3 кислоты, закрывают сосуд пробкой с мешалкой и термометром и производят 10 измерений температуры «предварительного периода» при равномерном перемешивании раствора. Через боковое ответвление реакционного сосуда вносят в него 25 см3 основания. При непрерывном перемешивании раствора производят измерения температуры в «главном» и «заключительном» периодах. Экспериментальные данные записывают в таблицу 1.
Таблица 1
| Электролиты (концентрация) | Температура, Т,°С | ||
| (предварительный период) | (главный период) | (заключительный период) | |
| KNO3 | 1. | 11. | 16. |
| 2. | 12. | 17. | |
| … | … | … | |
| 1. МеОН(0.5н) + +НА(0,5н) | 1. | 11. | 16. |
| 2. | 12. | 17. | |
| … | ... | … | |
| 2. ... | |||
| 3. ... | |||
Таблица 2
| Вариант | Задание | Вариант | Задание |
| 1. | NaOH + HCl (2н) | 9. | NaOH + HNO3 (0,5н) |
| NaOH + HAc (2н) | NH4OH + HNO3 (0,5н) | ||
| 2. | NaOH + HCl (1н) | 10. | KOH + HCl (2н) |
| NaOH + HAc (1н) | NH4OH + HCl (2н) | ||
| 3. | NaOH + HCl (0,5н) | 11. | KOH + HCl (1н) |
| NaOH + HAc (0,5н) | NH4OH + HCl (1н) | ||
| 4. | NaOH + H2SO4 (2н) | 12. | KOH + HCl (0,5н) |
| KOH + HAc (2н) | NH4OH + HCl (0,5н) | ||
| 5. | NaOH + H2SO4 (1н) | 13. | KOH + H2SO4 (2н) |
| KOH + HAc (1н) | NH4OH + H2SO4 (2н) | ||
| 6. | NaOH + H2SO4 (0,5н) | 14. | KOH + H2SO4 (1н) |
| KOH + HAc (0,5н) | NH4OH + H2SO4 (1н) | ||
| 7. | NaOH + HNO3 (2н) | 15. | KOH + H2SO4 (0,5н) |
| NH4OH + HNO3 (2н) | NH4OH + H2SO4 (0,5н) | ||
| 6. | NaOH + HNO3 (1н) | ||
| NH4OH + HNO3 (1н) |
 |
Обработка результатов
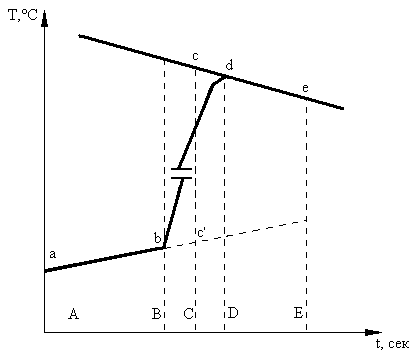
Рис.1. Зависимость изменения температуры во времени
На основании экспериментальных данных (табл.1) вычерчивают на миллиметровой бумаге график изменения температуры во времени (рис.1.). На рисунке АВ - предварительный, BD - главный, DE -заключительный периоды. Для графического определения точного значения ΔT проецируют точки В и D на ось ординат, находят середину отрезка BD и опускают перпендикуляр к оси ординат (сС). Продлевают линейные участки АВ и DE до пересечения с линией сС. Отрезок сс` соответствует изменению температуры ΔТ в калориметрическом опыте с учетом поправки на теплообмен.
Определим значение ΔТ для нитрата калия, вычисляют постоянную калориметра k по уравнению:
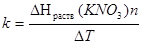 ,
,
где ΔHраств(KNO3) - известная энтальпия растворения нитрата калия, равная 36,02 (при 29 1 К) или 34,93 (при 298K) кДж/моль; n - количество вещества нитрата калия, моль.
Определяют энтальпию нейтрализации (ΔНi) для раствора данной i-той молярной концентрации (сi) по уравнению
ΔHi = kΔT
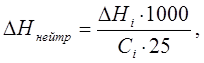 Вычислив энтальпию нейтрализации для данного количества кислоты или основания, рассчитывают интегральную энтальпию нейтрализации ΔHнейтр на один моль кислоты(основания).
Вычислив энтальпию нейтрализации для данного количества кислоты или основания, рассчитывают интегральную энтальпию нейтрализации ΔHнейтр на один моль кислоты(основания).
где 25 – объем раствора, см3.
Оценивают погрешность (П%) полученного значения энтальпии нейтрализации по отношению к табличному значению:
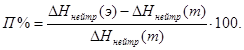
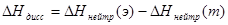 Учитывая, что при образовании 1 моль воды из ионов водорода и гидроксида выделяется 55,9 кДж, по закону Гесса определяют теплоту диссоциации уксусной кислоты или гидроксида аммония.
Учитывая, что при образовании 1 моль воды из ионов водорода и гидроксида выделяется 55,9 кДж, по закону Гесса определяют теплоту диссоциации уксусной кислоты или гидроксида аммония.
Результаты вычислений записывают втабл.3
Таблица З
| Электролит | С,моль/л | ΔT, °C | ΔH,КДж | ΔH нейтр , кДж/моль | 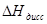 , кДж/моль , кДж/моль
|
| KNO3 | |||||
| NaOH+HCI | |||||
| NaOH+HAc | |||||
| (образец) |
Выводы
Исходя из экспериментальных и расчетных значений необходимо оценить силу изученных кислот и оснований, объяснить отклонение полученной экспериментально энтальпии нейтрализации от теоретической.
Лабораторная работа № 10
Исследование кинетики сорбции полимерами
компонентов агрессивной среды
Настоящие методические указания составлены в соответствии со стандартом по физической и коллоидной химии для студентов технических высших учебных заведений по разделам курса "Химическая кинетика» и «Коллоидная химия".
Цель работы: Исследование процессов сорбции агрессивных сред полимерами, сравнительная оценка агрессивности различных сред и химического сопротивления различных полимерных материалов.
Конструкционные полимерные материалы часто эксплуатируются в присутствии агрессивных сред. При этом протекают физические и химические процессы, сопровождающиеся изменением структуры и свойств полимеров. Различают физически и химически агрессивные среды.
Физически активной средой является та, которая не вступает в химическое взаимодействие с полимерами. Такие среды, как правило, вызывают обратимые изменения в полимерном материале, но иногда их воздействие приводит и к необратимым изменениям. Под воздействием физически активных агрессивных сред протекают процессы адсорбции (поверхностные процессы) и сорбции (поглощение в объеме) компонентов агрессивной среды полимерами. Адсорбция приводит к изменению значения поверхностной энергии на границе полимер – среда. Сорбция агрессивных сред вызывает, обычно, ослабление межмолекулярного взаимодействия в полимерах. Интенсивность воздействия физически активных сред на полимеры приближенно оценивается величиной сорбции, которая, согласно гильдебранту, будет тем больше, чем меньше разность параметров растворимости полимера и среды. В таблице 1 представлены параметры растворимости некоторых полимеров и растворителей.
Под действием на полимеры физически активных сред, например органических растворителей, может происходить их набухание. При набу3хании уменьшается прочность полимерного материала вследствие возникновения растрескивания полимеров, что обусловлено тремя причинами. Во-первых, высокая скорость набухания приводит к увеличению объема растворителя в образце полимерного материала. Во-вторых, растворитель, накапливаясь в трещинах и пустотах полимера, вызывает осмотические и капиллярные явления. В-третьих, агрессивная среда, проникая на границу раздела фаз наполнителя и связующего, уменьшает когезионную прочность. Предельным случаем набухания является растворение полимера.
Таблица 1
Параметры растворимости σ в (МДж/м3)1/2
| Полимеров | Растворителей |
| Политетрафторэтилен 12,7 | Диэтиловый эфир 15,5 |
| Полиэтилен 16,1 | Бутилацетат 17,4 |
| Полистирол 17,8 | Этилацетат 18,5 |
| Поливинилхлорид 19,8 | Хлороформ 19,0 |
| Полипропилен 16,7 | Ацетон 20,1 |
| Полиметилметакрилат 21,3 | Этанол 26,0 |
| Каучук 20,2 | Метанол 26,9 |
В отличие от физически активных, химически активные среды вызывают необратимые изменения химической структуры полимеров. Совокупность химических процессов, приводящих к изменению химической структуры полимера, его молекулярной массы под воздействием агрессивной среды, называется химической деструкцией. В этом процессе макромолекулы полимеров могут претерпевать распад основной цепи за счет:
· разрыва ковалентных связей и определяется законом случая или законом слабых связей;
· отщепления молекулы мономера от конца цепи макромолекулы – определяется законом концевых групп;
· превращение группы атомов в составе макромолекулы при сохранении исходной степени полимеризации;
· образование новых химических связей между макромолекулами, т.е. реакции сшивания, структурирования.
Химическая деструкция приводит к изменению массы полимерного материала и протекает либо на поверхности полимерного материала (внешняя диффузионно-кинетическая область), либо по объему изделия (внутренняя диффузионно-кинетическая или кинетическая область). При протекании деструкции во внешней диффузионно-кинетической области возможны два случая: если продукты деструкции растворимы в агрессивной среде, то масса полимерного изделия уменьшается значительно; если продукты деструкции практически не растворимы, то масса полимера будет уменьшаться в меньшей степени. При протекании деструкции во внутренней диффузионно-кинетической или кинетической областях, изменение массы будет зависеть от типа распада макромолекул и от способности продуктов деструкции растворяться в агрессивной среде. Если распад макромолекул происходит по закону случая, то масса полимерного изделия уменьшается незначительно. Если идет процесс деполимеризации макромолекул по закону концевых групп, то в случае растворения продуктов деструкции, масса полимера уменьшается в значительной степени. Очень часто процессы сорбции компонентов среды и деструкции происходят одновременно, что приводит к сложной зависимости изменения массы полимера от времени.
Реактивы и оборудование
Стаканы специальные, цилиндры мерные, весы аналитические, шкаф термостатический, спирт этиловый, набор агрессивных сред и образцы исследуемых полимерных материалов (по указанию преподавателя).
Опыт 1. Исследование кинетики сорбции агрессивной среды. Набухание полимеров.
1. Приготовить исследуемый раствор и залить его в стаканы.
2. Определить исходный объем (V0) всех образцов из двух разных материалов. Определение объема полимера следует производить в воде и спирте. В цилиндр налить около 20 мл дистиллированной воды или этанола. Затем в тот же цилиндр поместить образец полимера и зафиксировать изменение объема. Разность между начальным (V2) и конечным (V1) фиксированными объемами равна собственному объему исследуемого образца. (V0 = V2 – V1)
3. Осушить образец, промокнув фильтровальной бумагой, и погрузить его в агрессивный раствор.
Качественный анализ
1.1. ЛАБОРАТОРНАЯ РАБОТА №1
АНАЛИЗ СМЕСИ КАТИОНОВ I , II АНАЛИТИЧЕСКИХ ГРУПП
МЕТОДИЧЕСКИЕ УКАЗАНИЯ. Для выполнения лабораторной работы “АНАЛИЗ СМЕСИ КАТИОНОВ I, II АНАЛИТИЧЕСКИХ ГРУПП” необходимо знать основные понятия качественного анализа, классификацию катионов в сероводородном методе качественного анализа, теорию ионных равновесий, связанную с осаждением и образованием осадков, теорию осаждения, специфические реакции на катионы I и II аналитических групп.
КРАТКОЕ ТЕОРЕТИЧЕСКОЕ ВВЕДЕНИЕ. Химическая идентификация (обнаружение) – это установление вида и состояния фаз, молекул, атомов, ионов и других составных частей вещества на основе сопоставления экспериментальных и соответствующих справочных данных для известных веществ.
Вещества, используемые для проведения аналитических реакций, называют реагентами или реактивами. При использовании в качественном анализе ионных реакций в растворах (аналитических реакций), последние должны удовлетворять следующим требованиям: иметь легко наблюдаемый внешний эффект за счет изменения окраски раствора, образования осадка, растворения осадка или выделения газа; протекать достаточно быстро; быть практически необратимыми; обладать достаточной чувствительностью.
Чувствительность аналитических реакций характеризуется открываемым минимумом, минимальной (предельной) концентрацией и предельным разбавлением. Аналитическая реакция тем чувствительнее, чем меньше открываемый минимум и предельная концентрация и чем больше предельное разбавление.
Реакции обнаружения иона, выполняемые в растворе его чистой соли, называются индивидуальными. Индивидуальные реакции делят на характерные и общие.
Характерные реакции – реакции, в которых участвуют реактивы, взаимодействующие с небольшим числом ионов или с одним ионом.
Общие реакции – реакции, в которых участвуют реактивы, взаимодействующие со многими ионами. Строгой границы между общими и характерными реакциями нет.
Реактивы, дающие общие реакции, являются групповыми и служат для отделения одной группы ионов от другой.
Реактивы, взаимодействующие с небольшим числом ионов, называются селективными.
Реактивы, взаимодействующие с одним ионом, являются специфическими. Они позволяют обнаружить искомый ион в присутствии любых других ионов.
Целью работы является качественное определение состава раствора.
ВЫПОЛНЕНИЕ РАБОТЫ:
Раствор может содержать катионы: N Н4+, К+, N а +, Ва2+, Са2+.
Краткая схема хода анализа смеси катионов
I и II аналитических групп
1. Предварительные испытания: а) обнаружение иона аммония,
б) проба на присутствие катионов II группы.
2. Отделение катионов II группы от катионов I группы.
3. Анализ осадка: а) растворение осадка в СН3СООН, б) обнаружение Ва2+, в) осаждение Ва2+,г) обнаружение Са2+.
4. Анализ центрифугата: а) удаление NН4+, б) обнаружение К+,
в) обнаружение Nа+.
Проведение анализа смеси катионов I и II аналитических групп
1. Предварительные испытания:
а) Обнаружение иона аммония. К 1 капле исследуемого раствора прибляют 3 капли реактива Несслера. В присутствии ионов аммония образуется красно-бурый осадок (при очень малых количествах NН4+ раствор окрашивается в желто-бурый или желтый цвет).
б) Проба на катионы II группы. К 2 каплям анализируемого раствора прибавляют несколько капель (до щелочной реакции) 2 н. раствора гидроксида аммония, 1 каплю раствора хлористого аммония и 3 капли раствора карбоната аммония. В присутствии катионов II группы выпадает белый осадок. В этом случае катионы II группы отделяют от катионов I группы, так как они мешают обнаружению последних. При отсутствии осадка дальнейший анализ упрощается и проводится по п. 4.
2. Отделение катионов II группы от катионов I группы
К исследуемому раствору (около 1 мл) прибавляют несколько капель 2 н. раствора NH4OH до появления слабого запаха аммиака, не исчезающего при перемешивании раствора, и 2 капли 2 н. раствора NН4С1. Помещают пробирку с раствором в водяную баню и нагревают до 70°. К горячему раствору прибавляют 10 капель 2 н. раствора (NH4)2CO3, хорошо перемешивают и снова нагревают на водяной бане. Осадок центрифугируют и, не сливая раствора, проверяют полноту осаждения прибавлением 1 капли раствора (NH4)2CO3. Появление мути (осадка) означает, что полнота осаждения не была достигнута. В таком случае еще прибавляют 4—5 капель раствора (NH4)2CO3, снова центрифугируют и вновь проверяют на полноту осаждения. Добившись полноты осаждения, сливают раствор с осадка в другую пробирку.
В осадке — ВаСО3, СаСО3, в центрифугате — NН4+, К+, Nа +. Осадок исследуют по п. 3, центрифугат — по п. 4.
3. Исследование осадка
а) Растворение осадка. Осадок карбонатов (см. п. 2) растворяют в 10 каплях 2 н. раствора уксусной кислоты при нагревании на водяной бане и перемешивании стеклянной палочкой. В растворе—ионы Ва2+ и Са2+.
б) Обнаружение Ва2+. К 2 каплям полученного раствора прибавляют 3 капли раствора СН3СООNa и 2 капли раствора К2Сг2О7. В присутствии Ва2+ выделяется желтый осадок хромата бария ВаСгО4.
Центрифугируют осадок ВаСrО4, растворяют его в 1 капле концентрированной соляной кислоты и полученный раствор при помощи нихромовой проволоки вносят в бесцветное пламя горелки. Пламя окрашивается в желто-зеленый цвет, что подтверждает присутствие иона Ва2+.
в) Отделение Ва2+. Поскольку ион бария мешает обнаружению иона кальция, его удаляют. Для этого к раствору (см. п. 3, а) прибавляют 5 капель раствора CH3COONa и затем добавлять по каплям раствор К2Сг2О7, пока жидкость над осадком не окрасится в оранжево-желтый цвет. Раствор с осадком хорошо перемешивают, нагревают на бане (1—2 мин) и центрифугируют. Осадок отбрасывают, центрифугат исследуют на ион кальция.
г) Обнаружение Са2+. К полученному центрифугату (см. п. 3, в) прибавляют 5 капель раствора (NH4)C2O4. В присутствии иона кальция выпадает белый кристаллический осадок. Отделяют осадок от раствора и рассматривают его на дне пробирки.
Обнаружение иона Са2+ подтверждают ми-крокристаллоскопической реакцией.
Для этого помещают на предметное стекло 1 каплю раствора соли кальция, прибавляют к ней 1 каплю 2 н. раствора Н2SО4, смесь осторожно нагревают до начала образования кристаллов. После остывания пластинки наблюдают под микроскопом длинные игольчатые кристаллы СаSО4 • 2Н2О.
С помощью микрокристаллоскопической реакции нон кальция обнаруживают и в присутствии иона бария.
4. Исследование центрифугата:
а) Удаление NН4+ . При анализе центрифугата (см. п. 2) следует иметь в виду, что в нем содержатся в большой концентрации ионы аммония, введенные при отделении катионов II группы от катионов I группы. В присутствии же ионов аммония нельзя обнаружить ионы К+ и Nа+. Поэтому ионы аммония полностью удаляют из раствора. Для этой цели переносят исследуемый центрифугат в фарфоровый тигель (или чашку), укрепляют под тягой тигель в фарфоровом треугольнике и раствор осторожно выпаривают досуха. После выпаривания прокаливают тигель с остатком сухих солей до полного прекращения выделения белого дыма солей аммония.
Охлаждают тигель и делают пробу на полноту удаления. Для этого берут небольшую крупинку сухого остатка, растворяют в 2 каплях дистиллированной воды и прибавляют 2 капли реактива Несслера: отсутствие красно-бурого осадка служит признаком полного удаления NH4+. В случае неполного удаления иона аммония (образуется красно-бурый осадок) прокаливание продолжают до тех пор, пока проба с реактивом Несслера даст отрицательный результат.
Сухой остаток, свободный от солей аммония, растворяют в нескольких каплях дистиллированной воды (в том же тигле). Раствор разделяют на две части: одну исследуют на ион калия, другую — на ион натрия.
б) Обнаружение К+. К первой части полученного раствора (п. 4,а) прибавляют 2—3 капли раствора Nа3[Со(NО2)6] и дают постоять. Образование желтого кристаллического осадка свидетельствует о присутствии иона калия.
Проверочное обнаружение К+ проводится с тем же раствором по окрашиванию пламени в бледно-фиолетовый цвет, наблюдаемый через синее стекло. Для этого нихромовую проволочку смачивают в концентрированной соляной кислоте и прокаливают на пламени горелки (до полного исчезновения окраски пламени). Очищенную проволочку смачивают анализируемым раствором и снова вносят в бесцветное пламя. Наблюдают окраску пламени через синее стекло.
Ничтожные количества соединений натрия маскируют окраску пламени. Синее стекло поглощают желтые лучи и фиолетовое окрашивание будет заметно в присутствии солей натрия.
в) Обнаружение Nа+. Ион натрия обнаруживают с помощью реактива КН2SbО4 в нейтральной или слабощелочной среде.
Вторую часть (см. п. 4,а) проверяют на реакцию раствора и добиваются, чтобы она была нейтральной или слабощелочной. Затем осадок центрифугируют (если раствор не совсем прозрачен), к центрифугату прибавляют 2—3 капли раствора КН2SbО4 и охлаждают водопроводной водой. В присутствии ионов натрия выпадают белые кристаллики соли натрия, образование которых ускоряется при трении стеклянной палочкой стенок пробирки. С помощью микроскопа убеждаются, что образуемый осадок кристаллический. Аморфный осадок не является критерием присутствия в растворе иона Na+.
Проверочный способ - окрашивание пламени в яркий желтый цвет, не исчезающий в течение 10-15 секунд. Для этого нихромовую проволочку, смоченную исследуемым раствором, вносят в бесцветное пламя горелки.
2. КОЛИЧЕСТВЕННЫЙ АНАЛИЗ
2.1.Гравиметрический анализ
2.1.1.ЛАБОРАТОРНАЯ РАБОТА №2
Определение содержания сульфатов в питьевой воде
МЕТОДИЧЕСКИЕ УКАЗАНИЯ. Для выполнения лабораторной работы “Определение содержания сульфатов в питьевой воде гравиметрическим методом” необходимо знать теорию электролитической диссоциации, теорию ионных равновесий, связанную с осаждением и образованием осадков, теорию гравиметрического (весового) анализа.
КРАТКОЕ ТЕОРЕТИЧЕСКОЕ ВВЕДЕНИЕ. Питьевая вода, подаваемая населению централизованными системами водоснабжения, должна быть безопасной в эпидемиологическом отношении, безвредной по химическому составу и иметь благоприятные органолептические свойства (ГОСТ 2874-73). При наличии в воде веществ, придающих вкус (сульфаты, хлориды), сумма их концентраций, выраженная в долях от максимально допустимых концентраций каждого вещества в отдельности, не должна быть более 1.
Согласно ГОСТ 2874-73, химические свойства, влияющие на органолептические свойства воды, встречающиеся в природных водах или добавляемые процессе обработки, не должны превышать норм, указанных в таблице 1.
Таблица 1.
Концентрация химических веществ в воде (мг/л)
| Химическое вещество | Норма |
| Сухой остаток | 1000 |
| Хлориды | 350 |
| Сульфаты | 500 |
| Железо | 0.3 |
| Марганец | 0.1 |
| Медь | 1.0 |
| Цинк | 5.0 |
| Остаточный алюминий | 0.5 |
| Гексаметафосфат | 3.5 |
| Триполифосфат | 3.5 |
| Общая жесткость, мг-экв./л | 7.0 |
Содержание в питьевой воде повышенного количества сульфатов может оказать слабительное действие и изменить вкус воды. Качественное и количественное (мг/л) определение SO42- -ионов основано на учете степени помутнения воды от сульфата бария, образовавшегося при взаимодействии сульфат-иона с хлоридом бария: Ba2+ + SO42- ® BaSO4 ¯
Слабая муть, появляющаяся через несколько минут.…..1.0 -10
Слабая муть, появляющаяся сразу……….……………..….……10 - 100
Сильная муть……………………………………………………………..100 - 500
Большой осадок, быстро оседающий на дно…………….Более 500,
В насыщенном растворе малорастворимого электролита устанавливается равновесие между веществом в осадке и ионами его в растворе: BaSO4 Û Ba2+ + SO42 - ,
в осадке в растворе
константа равновесия которого 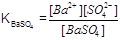
называется произведением растворимости ПР, т.к. в насыщенном растворе величина [BaSO4] при неизменной температуре является постоянной: [Ba2+][SO42-] = K [BaSO4] = const. Произведение растворимости при неизменной температуре и давлении - величина постоянная, количественно характеризующая способность данного электролита к растворению. В общем виде для малорастворимого электролита KaAb KaAb Û aK++ bA-
произведение растворимости выражается уравнением:
ПРKaAb = [K+]а [A-]b, где [K+] и [A-] - равновесные концентрации катионов и анионов, образующих при электролитической диссоциации электролита KaAb; a и b - стехиометрические коэффициенты в формуле у катиона и аниона.
Трудно растворимый электролит может оставаться в растворе до тех пор, пока произведение концентрации его ионов не превышает ПР. Если при смешивании растворов произведение концентраций ионов данного электролита окажется выше ПР, то избыточное количество электролита выпадает из раствора в виде осадка.
Процесс осаждения определяемого элемента из раствора в виде малорастворимого соединения лежит в основе гравиметрического метода химического анализа. Результаты гравиметрических определений обычно выражают в массовых процентах. Для вычислений в гравиметрическом анализе используют факторы пересчета F, называемые также аналитическими факторами. Фактор пересчета представляет собой отношение молярной массы определяемого вещества (М1) к молярной массе вещества, находящегося в осадке (М2): F = M 1 / M 2 . Фактор пересчета показывает, сколько граммов определяемого вещества содержит 1 г осадка.
Целью работы является ознакомление и овладение основными принципами и приемами гравиметрического метода анализа, определение содержания сульфатов в питьевой воде.
ВЫПОЛНЕНИЕ РАБОТЫ:
Качественная проба. В пробирку наливают 3 мл исследуемой воды, добавляют 0.5 мл соляной кислоты и 2 мл раствора хлорида бария. Образование осадка BaSO4 свидетельствует о присутствии в воде ионов SO42- .
Количественное определение. Пробу исследуемой питьевой воды объемом 100 мл фильтруют в колбу вместимостью 150 cм3 , в фильтрат добавляют 2-3 капли раствора метилового оранжевого и соляной кислоты до розовой окраски раствора. Смесь нагревают до кипения и выпаривают примерно наполовину на электроплитке. Дают отстояться раствору. При наличии мути или хлопьев фильтруют через беззольный фильтр. Фильтр промывают дистиллированной водой, фильтрат вместе с промывными водами вновь выпаривают наполовину. В кипящий раствор при помешивании приливают 10 мл горячего раствора хлорида бария. Раствор с осадком нагревают на водяной бане до осветления, проверяют полноту осаждения, прибавляя к прозрачному раствору 1-2 капли хлорида бария. Отсутствие мути указывает на полноту осаждения. Колбу накрывают часовым стеклом и нагревают на водяной бане в течение 20 минут, далее снимают и оставляют при комнатной температуре до охлаждения раствора.
Раствор фильтруют через плотный беззольный фильтр. Осадок BaSO4 несколько раз декантируют дистиллированной водой, отфильтровывая воду через беззольный фильтр. Осадок фильтруют через этот же фильтр, промывают горячей дистиллированной водой до тех пор, пока в фильтрате при добавлении раствора AgNO3 не будет появляться лишь небольшая муть. Фильтр с осадком аккуратно складывают в предварительно взвешенный тигель и помещают в муфельную печь. Прокаливают при доступе воздуха. Охлаждают в эксикаторе, взвешивают и определяют содержание сульфатов в исследуемом образце питьевой воды.
ОБРАБОТКА РЕЗУЛЬТАТОВ:
Содержание сульфатов (Х), мг/л, вычисляют по формуле
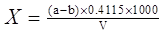 ,
,
где a - масса тигеля с осадком, мг; b - масса тигеля, мг; 0.4115 - коэффициент для пересчета BaSO4 на SO42-; V - объем воды, взятой для определения, мл.
2.2.Титриметрический анализ
2.2.1.Лабораторная работа №3
Определение качества хлебобулочных изделий
МЕТОДИЧЕСКИЕ УКАЗАНИЯ. Для выполнения лабораторной работы “Определение качества хлебобулочных изделий” по ГОСТ 5670-94 необходимо знать основы теории растворов и титриметрического метода анализа.
КРАТКОЕ ТЕОРЕТИЧЕСКОЕ ВВЕДЕНИЕ. Титриметрический анализ основан на законе эквивалентов и выполняется путем измерения количества реагента, необходимого для взаимодействия с определяемым компонентом в растворе. Для этого, раствор с точно известной концентрацией реагента (титрант) постепенно добавляют к раствору определяемого вещества, контролируя объем вводимого титранта. Процесс постепенного приливания раствора-титранта к раствору анализируемого вещества называют титрованием. В ходе титрования необходимо установить момент окончания реакции, т.е. определить точку эквивалентности. Точку эквивалентности устанавливают по изменению окраски индикатора (индикаторный способ), или других свойств раствора (физико-химические способы). При титровании вещества X стандартным раствором вещества A в точке эквивалентности количества эквивалентов этих веществ равны n(X) = n(A).
Под эквивалентом в данном случае понимается некая реальная или условная частица (молекула, ион, атом или их часть), которая в данной кислотно-основной реакции равноценна одному иону водорода. Молярная масса эквивалента - масса вещества, численно равная эквиваленту, выраженная в граммах.
При титровании концентрации стандартных растворов С(А) выражают, как правило, в эквивалентной (нормальной) концентрации (Сн) - количеством молей эквивалента (молярных масс эквивалента) растворенного вещества в 1 л раствора С(А) = n(А)/1000, где n(А) - количество молей эквивалента (эквивалентных масс) вещества А.
Раствор, содержащий 1 моль эквивалента вещества А в 1 л раствора, называется нормальным раствором (1н). Если известна нормальная концентрация стандартного раствора-титранта, то, учитывая, что вещества реагируют между собой в эквивалентных количествах, концентрация исследуемого раствора определяется как: C(X) V(X) = C(A) V(A)
Кислотность хлебобулочных изделий выражается в градусах кислотности.
Градусом кислотности называется объем в см3 1н раствора NaOH, необходимый для нейтрализации кислот, содержащихся в 100 г мякиша хлеба и хлебобулочных изделий. Кислотность вычисляют с точностью до 0.5 град.
В норме кислотность хлеба из пшеничной муки высшего и первого сорта должна быть не более 3 , второго сорта - 4 , кислотность ржаного хлеба не должна превышать 12 .
Целью работы является освоение основных приемов титриметрического анализа, определение качества хлебобулочных изделий на основании величины кислотности.
ВЫПОЛНЕНИЕ РАБОТЫ:
Из хлебобулочного изделия массой 200-500 г отрезают кусок массой около 70 г, у которого срезают корки и подкорочный слой около 1 см и, удалив все инородные включения, получают мякиш, который помещают в фарфоровый стакан, измельчают и перемешивают. Навеску 25 г измельченного мякиша переносят в сухой стакан вместимостью 400 см3 и приливают 50 мл дистиллированной воды с температурой около 600 С. Хлеб в стакане быстро растирают фарфоровой ложкой до получения однородной массы и добавляют еще 200 мл горячей дистиллированной воды. Полученную смесь аккуратно переносят в колбу на 500 см3. Колбу закрывают пробкой и энергично встряхивают в течение трех минут. После встряхивания дают смеси отстояться в течение одной минуты и отстоявшийся жидкий слой осторожно сливают в сухой стакан на 400 см3 через марлю. Из стакана отбирают пипеткой по 50 мл раствора в три конические колбы вместимостью по 100-150 см3 каждая и титруют 0.1 н раствором NaOH с 2-3 каплями фенолфталеина до получения слабо розового окрашивания, не исчезающего при перемешивании колбы в течение 1 минуты.
ОБРАБОТКА РЕЗУЛЬТАТОВ:
Кислотность (А) в градусах вычисляют по формуле
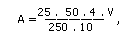
где V - объем 0.1 н раствора NaOH, см3; 1/10 - приведение 1 н раствора NaOH к 1 моль/дм3; 4 - коэффициент, приводящий к 100 г навески; 25 - масса навески испытуемого продукта, г; 250 - объем воды, взятый для извлечения кислот, см3; 50 - объем испытуемого раствора, взятого для титрования.
После титриметрического определения кислотности хлеба проводят статистическую обработку, полученные результаты сводят в таблицу:
| Ai | ni | `A | S | P при a=0.95 | D |
где Ai - объем щелочи, пошедшей на титрование, ni - число определений, `A - среднее значение; a - доверительная вероятность; ta - критерий Стьюдента; 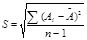 - стандартное отклонение;
- стандартное отклонение; 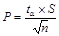 - правильность анализа;
- правильность анализа; 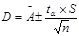 - доверительный интервал.
- доверительный интервал.
2.2.2.Лабораторная работа № 4.
Определение качества воды
МЕТОДИЧЕСКИЕ УКАЗАНИЯ. Для выполнения лабораторной работы «Определение качества воды» необходимо знатьосновы теории растворов и титриметрического метода анализа.
КРАТКОЕ ТЕОРЕТИЧЕСКОЕ ВВЕДЕНИЕ. Присутствие в воде гидрокарбонатов кальция и магния определяет ее жесткость. Жесткостью воды называется сумма миллиэквивалентов ионов Ca2+ и Mg2+ , содержащихся в 1 литре воды (мэкв/л). Различают временную жесткость, обусловленную содержанием гидрокарбонатов, и постоянную, связанную с присутствием других солей Mg и Ca (сульфаты, хлориды).
Принято считать воду с жесткостью более 6,5 мэкв жесткой; 6,5-3 мэкв - средней жесткости, ниже этой величины – мягкой.
Определение временной (карбонатной) жесткости основано на взаимодействии ионов Ca 2+ и Mg2+ , находящихся в воде в виде гидрокарбонатов, с соляной кислотой по реакциям
Ca(HCO3)2 + 2HCl = CaCl2 + 2H2O + 2CO2
Mg(HCO3)2 + 2HCl = MgCl2 + 2H2O + 2CO2
По объему соляной кислоты известной концентрации, пошедшей на взаимодействие с растворенными в воде солями, обусловливающими временную жесткость, рассчитывают содержание последних в исследуемой воде.
Определение временной жесткости проводится методом титрования. Для определения окончания реакции используют индикатор метилоранж, изменяющий окраску с желтой на оранжевую при появлении в исследуемом растворе небольшого избытка кислоты.
Определение общей жесткости основано на способности двузамещенной натриевой соли этилендиаминтетрауксусной кислоты ЭДТА (трилон Б) образовывать с ионами магния и кальция малодиссоциированные комплексы по реакции:

|
из
5.00
|
Обсуждение в статье: Определение энтальпии нейтрализации |
|
Обсуждений еще не было, будьте первым... ↓↓↓ |

Почему 1285321 студент выбрали МегаОбучалку...
Система поиска информации
Мобильная версия сайта
Удобная навигация
Нет шокирующей рекламы