 |
Главная |


Другие реакции присоединения алкенам
|
из
5.00
|
v Присоединение серной кислоты (сульфатирование). Алкеныреагируют на холоду с концентрированной серной кислотой с образованием кислых алкилсульфатов общей формулы R-OSO3H. Эти вещества образуются в результате присоединения протона к одному из атомов углерода, связанного двойной связью, и гидросульфат-иона – к другому. Важно отметить, что углерод образует связь с кислородом, а не с серой.
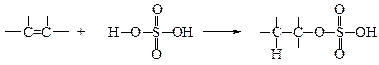
Если раствор кислого алкилсульфата в серной кислоте разбавить водой и нагреть, то образуется спирт, имеющий ту же алкильную группу что и исходный кислый алкилсульфат. Кислые алкилсульфаты разлагаются водой с образованием спирта и серной кислоты, т.е. происходит гидролиз. Присоединение серной кислоты к алкенам проводят с целью получения спиртов.



Это хороший промышленный метод синтеза спиртов, поскольку алкены сравнительно легко получаются при крекинге нефти. Присоединение серной кислоты протекает по правилу Марковникова, и поэтому первичные спирты, кроме этилового, нельзя получить этим методом. Свойства алкенов растворяться в серной кислоте используются и для очистки их от алканов и галогеналканов, которые в последней нерастворимы.
v Присоединение воды (гидратация). Реакция гидратации алкенов протекает в присутствии минеральных кислот, чаще всего серной.
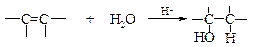
Механизм реакции аналогичен механизму присоединения галогеноводородов.
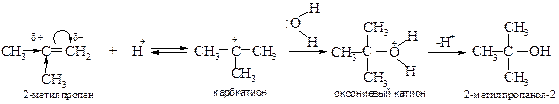
Лимитирующей стадией реакции является образование карбкатиона. Эта стадия обратима, как и весь процесс гидратации. Важно, что взаимодействие карбкатиона с водой при ее избытке идет быстрее, чем обратная реакция регенерации алкена. Наряду с реакцией карбкатиона с водой протекает его конкурирующая реакция с анионом серной кислоты.
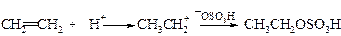
Образовавшийся этилсульфат при нагревании гидролизуется до этилового спирта.
При гидратации несимметричных алкенов реакция протекает по правилу Марковникова, а гидратация разветвленных алкенов часто сопровождается перегруппировкой. Так, например, из 2,2-диметилбутена-1 при гидратации образуется главным образом 2,3-диметилбутанол-2. Первоначально возникающий при протонировании алкена вторичный карбкатион перегруппировывается в более стабильный третичный карбкатион (алкильный сдвиг), который и атакует молекула воды.
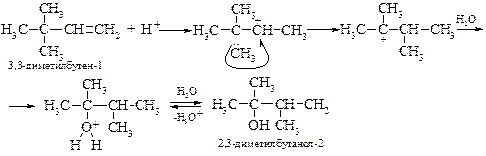
§ В последние годы разработан более эффективный промышленный метод гидратации в присутствии менее агрессивной (и менее нуклеофильной) фосфорной кислоты в смеси с вольфрамовой кислотой, нанесенной на силикагель (давление 100 атм. и температура 300-350 ºС). В этом случае образующийся карбкатион реагирует только с водой с образованием спирта.
Реакцию гидратации можно также осуществить и в газовой фазе (повышенная температура и давление). В качестве катализаторов применяют оксид алюминия, хлорид цинка и др.
Поскольку такое присоединение происходит по правилу Марковникова, то получаются те же спирты, что и в предыдущей реакции. Такая непосредственная гидратация – наиболее простой и дешевый из двух способов. Гидратация широко используется в промышленности для синтеза трет-бутилового спирта из изобутилена и трет-амилового (2-метилбутанола-2) – из триметилэтилена.
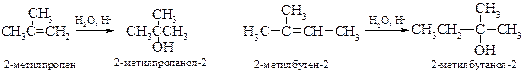
v Присоединение ацетата ртути (оксимеркурирование). Обработка алкенов ацетатом ртути в воде, спиртах или карбоновых кислотах с последующим восстановлением боргидридом натрия приводит соответственно к спиртам, простым и сложным эфирам. Более подробно метод рассмотрен в главе 13 «Спирты».
v Получение галогенгидринов (гипогалогенирование). Галогенгидрины – соединения с вицинальным расположением гидроксильной группы и атома галогена.
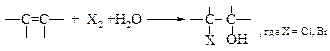
Этилен реагирует с бромом в присутствии воды с образованием этиленбромгидрина. Аналогично реагируют и другие алкены. Первоначально под действием катиона брома происходит образование промежуточного карбкатиона, который реагирует с молекулой воды.
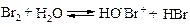
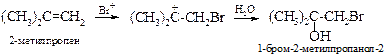
v Реакция Принса[13]. Алкены присоединяют формальдегид в кислой среде по следующему механизму. Первоначально формальдегид подвергается протонизации.
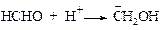
Образовавшийся катион атакует олефин по двойной связи, образуя новый карбкатион, который в присутствии воды и кислых катализаторов (H2SO4, H3PO4, BF3, ZnCl2) превращается в 1,3-диол.
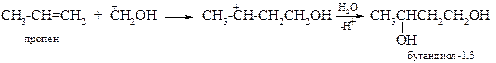
Важность этой реакции заключается в том, что такой диол легко превращается в бутадиен-1,3 СН2=СН-СН=СН2.
В другом случае в безводной среде из 2-метилпропена при температуре около 200 ºС образуется непредельный спирт – 3-метилбутен-3-ол-1, который может быть превращен в изопрен.

v Реакция гидроформилирования алкенов (реакция Реппе[14]). Одним из наиболее эффективных методов введения в олефины карбонильной группы является синтез альдегидов в присутствии комплексов переходных металлов в токе водорода.
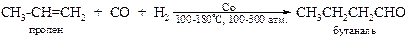
Считают, что катализатором служит образующийся в этих условиях тетракарбонилкобальтат водорода Н[Co(CO)4]. На сегодня это один из наиболее эффективных промышленных методов получения альдегидов.
Замена катализатора на комплекс пентакарбонилкобальтата с трифенилфосфином позволяет на порядок снизить давление, но при этом альдегиды восстанавливаются до спиртов.
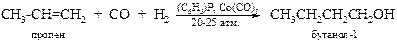
v Димеризация и алкилирование. В главе 7 «Алканы» обсуждались реакции димеризации алкенов и конденсации алкенов с алканами, протекающие в присутствии кислотных катализаторов. Реакции идут через образование карбкатионов. Например:
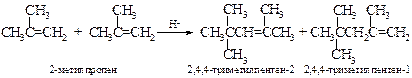

v Гидроборирование-окисление. Один из наиболее эффективных методов получения спиртов. Более подробно будет рассмотрен в главе 13 «Спирты».

v Свободно-радикальное присоединение тетрагалогенметанов. В условиях образования свободных радикалов тетрагалогенметаны присоединяются по двойной связи. Данная реакция лежит в основе получения b-гидроксикарбоновых кислот.
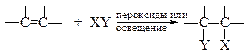
| Например: | 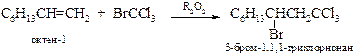
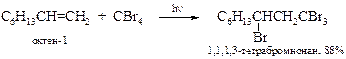
|
v Присоединение карбенов. В предыдущей главе подробно рассматривалось образование карбенов и их реакции с алкенами, приводящие к образованию производных циклопропана.
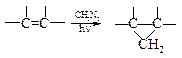
| Например: | 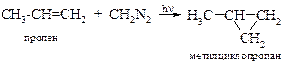
|
Реакции замещения
v Аллильное галогенирование. Поскольку алкильные группы в олефинах имеют структуру алканов, они должны вступать в реакции замещения.
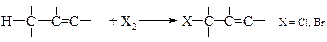
Если необходимо провести галогенирование алкильной части молекулы алкена, то реакцию проводят в условиях свободно-радикального замещения (механизм SR).
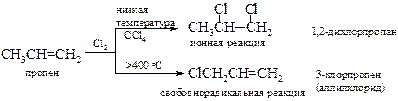
Хлорирование пропилена в аллильное положение (аномальное хлорирование) при высокой температуре впервые наблюдал М. Львов[15] в 1883 г.
Высокая температура требуется для протекания реакции замещения с низкомолекулярными алкенами с неразветвленной цепью, тогда как для олефинов с разветвленной цепью могут протекать и при комнатной температуре.
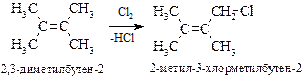
Принципиально в ходе высокотемпературного хлорирования может происходить присоединение по двойной связи радикала хлора с образованием первичного и вторичного алкильных радикалов, а также свободно-радикальное замещение атома водорода в метильной группе. Второй процесс превалирует, так как, аллильный радикал более устойчив, а химическая реакция, как известно, протекает через переходное состояние с меньшей энергией активации, которому и соответствует более устойчивый интермедиат.
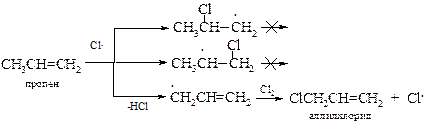
· Аллильный радикал. Стабильность промежуточного аллильного радикала СH2=CH‑СH2× объясняется р,p-сопряжением электронов двойной связи с р-орбиталью неспаренного электрона, что приводит к делокализации электронной плотности и выравниванию связей.
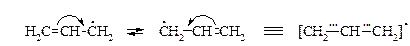
Подтверждением высокой стабильности аллильного радикала может служить уменьшение энергия разрыва связи С-Н в аллильном положении по сравнению со связью С-Н в насыщенном углеводороде – 364 и 402 кДж/моль соответственно.
При рассмотрении структуры аллильного радикала методом МО видно, что первая связывающая орбиталь Y1 охватывает все три атома углерода. Вторая МО (Y2) – несвязывающая, имеет одну узловую плоскость и демонстрирует делокализацию свободного электрона по концевым атомам углерода. Третья разрыхляющая МО (Y3) имеет две узловые плоскости.
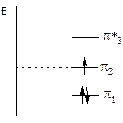

Рис. 8.5. Энергетические уровни и очертания АО и МО аллильного радикала
§ Бромирование в аллильное положение можно провести в мягких условиях под действием N-бромсукцинимида, который создает постоянную, но небольшую концентрацию брома.
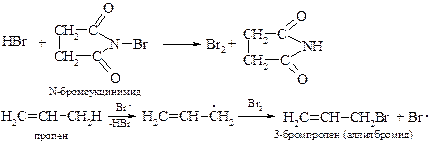
Низкая концентрация брома исключает реакцию электрофильного присоединения галогена по двойной связи алкена. Примененный впервые в 1942 г. К. Циглером этот вариант радикального бромирования получил широкое распространение в органическом синтезе.
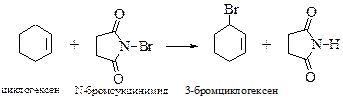
v Окислительный аммонолиз (аммоксидирование). Сравнительная легкость образования аллильного радикала используется в промышленности при окислительном аммонолизе пропилена с целью получения акрилонитрила.
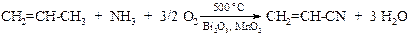
v Высокотемпературное хлорирование этилена. Замещение водорода на хлор в этилене при кратковременном (~2 сек) нагреве реакционной смеси до высокой температуры приобрело важное промышленное значение. Вполне возможно, что сначала в реакции образуется 1,2-дихлорэтан, который в результате элиминирования хлороводорода превращается в винилхлорид.
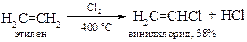
Реакции окисления
В отличие от алканов алкены легко окисляются прежде всего по двойной связи. Глубина окисления зависит от типа окислителя и условий проведения реакции.
v Окисление до эпоксидов. Окисление алкенов 25% пероксидом водорода в уксусной кислоте дает эпоксиды (a-окиси). Реакция идет через образование надуксусной кислоты.

Н. Прилежаевым[16] показано, что для получения a-окисей можно использовать готовые надкислоты. Предполагается, что реакция протекает через циклическое переходное состояние.
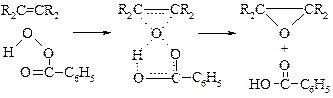
Ранее для эпоксидирования алкенов в основном использовали м-хлорпероксибензойную кислоту, которую сейчас в лабораторных синтезах заменила магниевая соль монопероксифталевой кислоты.
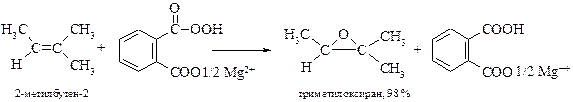
§ Олефины можно окислять до эпоксидов также с помощью полимерного материала – смолы, содержащей карбоксильные группы и обработанной пероксидом водорода. Прореагировавшая смола легко регенерируется.
§ В промышленности этиленоксид (оксиран) получается окислением этилена воздухом в присутствии серебряного катализатора.
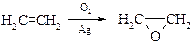
v Окисление до гликолей. Холодный разбавленный раствор перманганата калия (реакция Вагнера[17], 1888 г.) превращает олефины в гликоли.
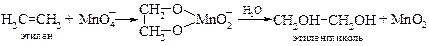
Реакция Вагнера имеет в настоящее время незначительное синтетическое значение из-за ее неселективности. Однако она может использоваться как качественная проба на двойную связь (обесцвечивание перманганата).
Изучение механизма окисления алкенов с применением перманганата калия, меченного изотопом 18О, показало, что источником обоих атомов кислорода в гликоле является перманганат-анион. Реакции носят стереоселективный характер и представляют собой цис-присоединение гидроксильных групп по двойной связи.
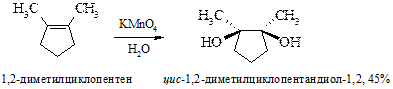
§ В настоящее время для гидроксилирования алкенов чаще применяют оксид осмия(VIII) (реакция Криге, 1936 г.). Преимущество перед реакцией Вагнера состоит в возможности в качестве растворителей использовать органические соединения – эфир, бензол, хлороформ. Реакция ускоряется пиридином. Так же как и в предыдущем случае образуются цис-вицинальные гликоли. В этой реакции требуется эквимольное количество довольно дорогого реагента. Поэтому вместо реакции Криге используют реакцию Майласа, где требуются каталитические количества оксида осмия совместно с пероксидом водорода.
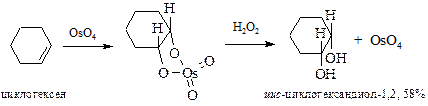
§ анти-1,2-Гидроксилирование алкенов осуществляют действием 30% пероксида водорода в уксусной кислоте в присутствии каталитических количеств серной кислоты.
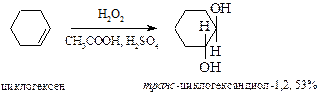
§ В органическом синтезе все чаще применяют окисление олефинов в неводной среде тетраацетатом свинца, которое приводит к диацетильным производным гликолей. Последние можно дезацетилировать до диолов.
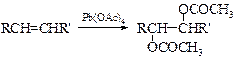
v Окисление жесткими окислителями. При действии концентрированных растворов сильных окислителей (KMnO4, HNO3, CrO3, K2Cr2O7) молекула алкена расщепляется по месту двойной связи, образуя кислоты и кетоны. Концевая метиленовая группа окисляется до углекислого газа.
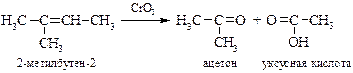
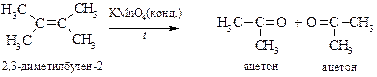
В синтетических целях реакция используется редко из-за низких выходов целевых продуктов. Однако этот недостаток может быть устранен, если реакцию окисления перманганатом калия проводить в бензоле в присутствии краун-эфира или добавляя оксид рутения(IV). Существуют реагенты, позволяющие остановить окисление альдегидов до кислот, например хромилхлорид CrO2Cl2.
Данные реакции до появления спектральных методов использовались для установления строения алкенов. Анализируя структуру продуктов окисления можно установить как число углеродных атомов в олефине, так и место расположения двойной связи, а также степень ее замещения, например:

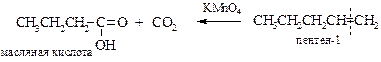
§ Реакция Лемье. Действуя на олефины иодной кислотой HIO4 и каталитическим количеством перманганата калия, Р. Лемье[18] провел следующую реакцию расщепления двойной связи:
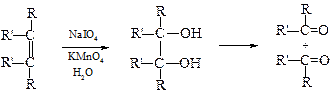 .
.
v Озонолиз. Аналогичные результаты можно достичь при окислении алкенов озоном по Гарриесу[19]. Озон присоединяется по месту двойной связи, образуя нестойкие, взрывчатые озониды, которые сразу разлагают водой до альдегидов или кетонов. Образующийся при этом пероксид водорода окисляет альдегиды до карбоновых кислот. Если же разложение озонидов вести водой в кислой среде в присутствии цинковой пыли, то окисление альдегидов не происходит.
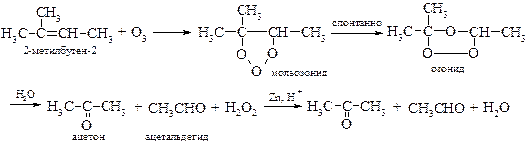
Если озонид обрабатывают боргидридом натрия NaBH4, то в качестве конечных продуктов выделяют смесь соответствующих спиртов.

Реакция озонирования была успешно использована К. Гарриесом в установлении строения каучука, в результате которой был получен единственный продукт реакции ‑ левулиновый альдегид, что можно представить схематически следующим образом:
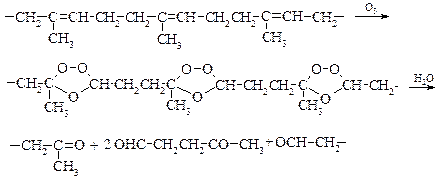
v Каталитическое окисление этилена (Ваккер-процесс). К реакциям окисления относится гладкое превращение этилена в ацетальдегид.

Предполагаемый механизм реакции:
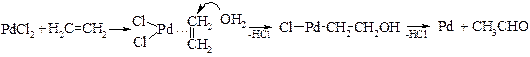 .
.
Хлорид меди(II) играет роль окислителя палладия.
2CuCl2 + Pd → PdCl2 + 2CuCl
Образующийся хлорид меди(I) вновь окисляется кислородом до хлорида меди(II). В настоящее время она является основным источником получения ацетальдегида в промышленности.
В заключение следует отметить, что важные для алкенов реакции полимеризации детально рассматриваются в главе "Диеновые углеводороды".
Важнейшие алкены
Наиболее широкое применение нашли первые представители гомологического ряда алкенов – этилен и пропен (пропилен). Промышленное применение этих соединений представлено далее на схемах.

Рис. 8.6. Схема промышленного использования этилена

Рис. 8.7. Схема промышленного использования пропилена
[1] Циглер К. (1998-1973) – немецкий химик. Основные работы в области органического синтеза и химии полимеров. Совместно с Д. Натта – Нобелевский лауреат (1963 г.).
[2] Натта Д. (1903-1973) – итальянский химик. Основные труды в области полимеров.
[3] Зайцев А. (1841-1910) – русский химик-органик, ученик А. Бутлерова. Исследования в области органического синтеза и теории химического строения.
[4] Гофман А. (1818-1892) – немецкий химик-органик. Основные работы в области азотсодержащих органических соединений. Предложил способы получения аминов, в том числе, из алкилгалогенидов (реакция Гофмана), разложением амидов (перегруппировка Гофмана).
[5] Реформатский C. (1864 – 1937) – украинский химик. Основные интересы сосредоточены в области химии природных соединений и металлорганического синтеза.
[6] Чугаев Л. (1873-1922) – русский химик. Главная область интересов – химия комплексных соединений. Установил повышенную устойчивость комплексов, образующих пяти- и шестичленные циклы (правило Чугаева), открыл качественную реакцию на никель (реактив Чугаева), метод количественного определения подвижных атомов водорода (метод Чугаева – Церевитинова).
[7] Берч А. (1915-1995) – австралийский химик. Основные работы посвящены непредельным соединениям и природным веществам.
[8] Виттиг Г. (1897 – 1987) – немецкий химик. Основные работы посвящены синтезу сложных органических соединений. Нобелевская премия 1979 г.
[9] Адамс Р. (1889-1971) – американский химик-органик. Основные исследования связаны с химией биологически активных соединений.
[10] Уилкинсон Дж. (р.1921) – английский химик. Исследования в области элементорганической химии, в частности, фероцена и других сэндвичевых соединений. Предложил гомогенный катализатор реакций гидрирования. Нобелевская премия 1973 г.
* Утверждение, что та или иная реакция является электрофильной или нуклеофильной, всегда относится к реагенту, т.е. к веществу, более простому по своей структуре.
[11] Марковников В. (1837-1904) – русский химик-органик. Исследования в области теоретической органической химии, органического синтеза и нефтехимии.
[12] Караш М. (1895-1957) – американский химик-органик. Основная область исследований – химия свободных радикалов.
[13] Принс Г. (1899-1958) – нидерландский химик-органик. Открыл реакции присоединения формальдегида к алкенам и галоформов или тетрагалогенметанов к полигалогенолефинам, названные его именем.
[14] Реппе В. (1892-1969) – немецкий химик. Основные работы посвящены химии ацетилена. Открыл ряд реакций, носящих его имя.
[15] Львов М. (1848-1899) – русский химик-органик. Ученик А. Бутлерова. Основные работы в области теории органического строения, в частности впервые получил неопентан, и химии непредельных соединений.
[16] Прилежаев Н. (1877-1944) – русский, советский химик-органик. Основные работы посвящены кислородсодержащим соединениям.
[17] Вагнер Е. (1849-1903) – русский химик-органик. Исследования в области органического синтеза и химии терпенов.
[18] Лемье Р. (1920-2000) – канадский химик. Исследования в области теоретической и синтетической химии углеводов.
[19] Гарриес (Харриес) К. (1866-1923) – немецкий химик. Основной объект изучения – реакции непредельных соединений, в т.ч. каучука.
|
из
5.00
|
Обсуждение в статье: Другие реакции присоединения алкенам |
|
Обсуждений еще не было, будьте первым... ↓↓↓ |

Почему 1285321 студент выбрали МегаОбучалку...
Система поиска информации
Мобильная версия сайта
Удобная навигация
Нет шокирующей рекламы