 |
Главная |


Наиболее распространенные митохондриальные болезни
|
из
5.00
|
| Болезнь | Дефектные белки |
| Болезнь Альцгеймера | ПируватДГ, a-кетоглутарат ДГ, белки комплекса IV ЭТЦ |
| Синдром Барта | АТФаза |
| Болезнь Паркинсона | Белки комплексов I, II, III ЭТЦ |
| Синдром Кернса-Сейера | Цитохром b |
| Атаксия Фридрейха | Белки комплексов I, II, III ЭТЦ |
| Синдром внезапной смерти младенца | АцилКоА- ДГ b-окисления ВЖК |
| Врожденная оптико-нейропатия Лебера (morbus Leber) | Белки комплексов I, IV ЭТЦ |
| Семейная гипертрофическая кардиомиопатия | Митохондриальные РНК |
| Болезнь Гентингтона | Белки комплексов I, II, III ЭТЦ |
| Синдром Лейга (Лея) | ПируватДГ |
| Мигрень | Белки кальциевых каналов |
| Семейный боковой амиотрофический склероз | СОД |
Нет ни одной отрасли клинической медицины, в которой бы не были описаны те или иные синдромы, связанные с патологией митохондрий: в эндокринологии – сахарный диабет, гипофизарный нанизм; в педиатрии – витамин Д-резистентный рахит, синдром «внезапной смерти младенца», в кардиологии – кардиомиопатии; в дерматологии – пятнистая пигментация кожи; в офтальмологии – ретинопатии и т.д.
У людей с одной и той же мутацией могут проявляться любые разнообразные симптомы от их отсутствия до тяжелых нарушений, а также развиваться самые различные болезни. Подобная разнохарактерность клинических проявлений объясняется присутствием как нормальных, так и мутантных митохондриальных хромосом в разных пропорциях в разнообразных тканях.
Для митохондриальной наследственности характерны следующие признаки. Данные заболевания передаются только по материнской линии, т.к. при слиянии гамет мутантная ДНК попадает в зиготу только из яйцеклетки. Пораженные девочки, выходящие замуж, будут рожать только больных детей, в то время как у больных мужского пола все потомство будет свободно от данного недуга (рис. 8).
Академик В.П. Скулачев считает, что именно митохондрии ответственны за феноптоз – запрограммированную смерть организма как закономерный результат возрастных изменений. Недавние исследования подтвердили, что окислительные повреждения мДНК могут быть главной причиной физического старения.
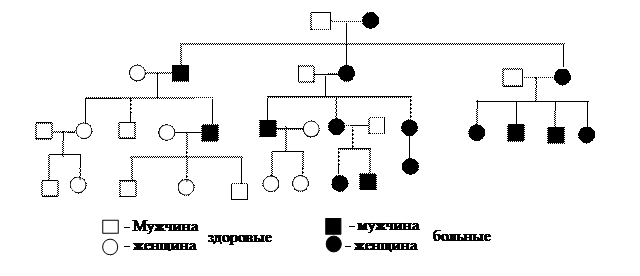
Рис. 8. Родословная, иллюстрирующая передачу дефектного признака через митохондрии.
Общими симптомами для подавляющего большинства митохондриальных болезней служат замедление нервно-психического развития, нарушения в деятельности мышечной системы, задержка роста, в основе которых лежат дефекты структуры митохондрий и их функций, обусловливающие энергетическую недостаточность клеток, в первую очередь, зависящих от интенсивности аэробных реакций (окислительного декарбоксилирования ПВК, ЦТК, ЭТЦ, окислительного фосфорилирования). Большое семейство генов аутосом, Х-хромосомы, митохондрий, ответственных за биоэнергетические процессы, получило наименование oxphos-генов (генов окислительного фосфорилирования). При наследственных болезнях, связанных с их мутациями, наиболее тяжело поражаются органы, требующие максимального обеспечения энергией (развивающийся мозг, миокард, скелетная мускулатура), поэтому устойчивость энергетического метаболизма гарантируется двойным генным контролем (ядерной и митохондриальной ДНК).
В 1951 году D. Leigh (Лей) описал патологию, первые признаки которой проявляются на 1-3 годах жизни (синдром Лея,syndrome Leigh).
В его основе генетический дефект тимидиндифосфаткиназы, осуществляющей преобразование:
 |
Имеет аутосомно-рецессивный тип наследования. Среди основных признаков: симптомокомплекс «вялого ребенка», задержка психомоторного развития, сниженная масса тела, анорексия, частая рвота. Позднее присоединяются гипотония или гипертонус мышц, судороги, тремор конечностей, расстройство координации, атаксия, сонливость, респираторный дистресс-синдром, нистагм, атрофия зрительных нервов, потеря слуха, нарушение дыхательной вентиляции (апноэ). Среди биохимических показателей – лактатацидоз (повышенные концентрации лактата, пирувата в крови и ликворе), уменьшение величин карнитина, активности цитохромоксидазы в клетках культуры тканей (фибробластах). Обычно через несколько лет недуг заканчивается летальным исходом, вследствие паралича дыхательного центра, так как попытки лечения не эффективны.
Синдром Барта(syndrome Bart), описанный в 1983 году, проявляется мышечной гипотонией, дилатационной кардиомиопатией, пропорциональной задержкой роста, нейтрофилопенией, прогрессирующей энцефалопатией. Наследуется по Х-сцепленному рецессивному типу, дефектный ген локализуется на длинном плече Х-хромосомы, в норме отвечает за синтез 3-гидроксиацилКоА-ДГ. Поэтому нарушается рН крови (ацидоз), развивается ацидурия (за счет увеличения содержания 3-метилглукановой, 3-метилглутаровой кислот – продуктов повреждения окисления лейцина). Попытки терапии (стимуляция активности ЭТЦ с помощью введения карнитина, КоQ, пантотеновой кислоты) могут иметь положительный, но кратковременный эффект.
Болезнь Паркинсона (morbus Parkinson) (табл. 2) имеет позднее начало. Ее первые симптомы появляются в 40-70 лет. В основе лежат симптомы преждевременной инволюции экстрапирамидной системы: а) двигательные расстройства (акинезия, ригидность мышц, маскообразное лицо, мелкие скользящие шаги, монотонная речь, дрожание рук и головы; б) вегетативные поражения (гипергидроз, гиперсаливация); в) психические нарушения (депрессия, позднее нарушается интеллект, галлюцинации).
Наследуется по аутосомно-доминантному типу. Частота 150-300:100000 населения. Если болезнь развивается в возрасте до 45 лет, то такие случаи принято обозначать термином ювенильной (ранний) паркинсонизм. Это особый недуг, аутосомно-рецессивный, часто причиной служит дефект гена, локализованного на хромосоме 6q и кодирующего синтез белка–паркина, который является важнейшим звеном системы клеточной защиты. При повреждении его образования гибнут нейроны в черной субстанции, что клинически проявляется пирамидными нарушениями, мышечной ригидностью.
Среди механизмов, предрасполагающих к паркинсонизму, следует выделить мутации в митохондриальной ДНК, в комплексе I ЭТЦ, в генах белков репаративной системы.
Обе формы диагностируются в основном по клиническим симптомам. Лечение симптоматическое.
Митохондриальная природа болезни(атаксии) Фридрейха (ataxia Friedreich) была доказана в 1996 году, когда был идентифицирован ген FRDA, мутации в котором и приводят к заболеванию. Наследуется по аутосомно-рецессивному типу. Данный транскриптон кодирует белок фратаксин, наиболее высокая экспрессия которого характерна для миокарда, скелетной мускулатуры, печени, поджелудочной железы, спинного мозга. У больных уровень этого белка в митохондриях резко снижен, что уменьшает содержание железа, так как фратаксин регулирует его транспорт в этих органеллах. В результате дефекта гена FRDA угнетается резистентность митохондрий к оксидантному стрессу и окислительным повреждениям, снижается скорость окислительного фосфорилирования – страдает энергетический метаболизм клетки. Болезнь проявляется сочетанием неврологических и экстраневральных нарушений (мозжечковой атаксией, слабостью мышц спины, ног, дизартрией, деформацией скелета, эндокринопатиями, поражением миокарда, атрофией дыхательного нерва, снижением IQ.
Для болезни Кернса-Сейера (morbus Cairns-Saier) характерно начало в детском возрасте. Основная симптоматика: прогрессирующая офтальмоплегия, ретинит, миопатия, атаксия, глухота, умственная отсталость, низкий рост, нарушение сердечной проводимости. В крови лактатацидоз. При ДНК-диагностике выявляются крупные делеции мДНК.
Среди биохимических исследований особое значение имеет регистрация системного лактатацидоза, а также изучение активности митохондриальных дегидрогеназ (в лимфоцитах). Распространенность атаксии 1:50000 населения. Благоприятно влияют на функционирование поврежденных систем различные антиоксиданты (витамин Е, коэнзим Q10 и др.).
Таким образом, наследственная патология биоэнергетики может быть обусловлена мутациями митохондриальных генов или сочетанным повреждением ядерных и митохондриальных транскриптонов, относительно часто несовместимыми с жизнью.
Для диагностики митохондриальных болезней решающее значение имеют биохимические, молекулярно-генетические, гистохимические, хроматографические, спектрофотометрические и другие методы изучения структуры и функций митохондрий биоптатов тканей или клеток крови. Принципы терапии подобных заболеваний аналогичны способам коррекции любой наследственной патологии.
|
из
5.00
|
Обсуждение в статье: Наиболее распространенные митохондриальные болезни |
|
Обсуждений еще не было, будьте первым... ↓↓↓ |

Почему 1285321 студент выбрали МегаОбучалку...
Система поиска информации
Мобильная версия сайта
Удобная навигация
Нет шокирующей рекламы