 |
Главная |


Выбор метода анализа ключевого компонента.
|
из
5.00
|
При диссоциации ключевого компонента образуется два иона:

Т.е. возможно количественное определение ключевого компонента по одному из этих ионов. При этом следует учесть тот факт, что образующийся побочный продукт реакции, тоже диссоциирует:
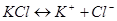
Т.е. в растворе присутствуют катионы калия от обеих солей, поэтому раздельное количественное определение их по катиону невозможно. Остаётся анион йода, а т.к. расходующийся на реакцию йод надёжно связан с метильным радикалом, он не будет присутствовать в растворе и мешать определению непрореагировавшего количества анионов йода, а т.к. диссоциация идёт в мольном соотношении, то следовательно и количества непрореагировавшего ключевого компонента.
Анион йода, при совместном присутствии аниона хлора, наиболее быстро, надёжно и относительно дёшево можно определить осадительным титрованием, а, в частности, аргентометрией [5].
Но для повышения точности анализа вследствие замены недостаточно точных визуальных индикаторов на показания, которые фиксирует прибор, наилучшим методом определения в данном случае будет потенциометрическое осадительное титрование [6].
Теоретические основы метода. Потенциометрический метод анализа основан на измерении потенциала электрода, опущенного в исследуемый раствор, и нахождении зависимости между его величиной и концентрацией определяемого компонента в растворе.
Практически невозможно измерить потенциал одного электрода, поэтому составляют гальванический элемент:
Измерительный | Исследуемый раствор | Электрод сравнения
(индикаторный) электрод | |
И измеряют э.д.с. элемента, то есть разность потенциалов двух электродов, опущенных в исследуемый раствор: э.д.с. = E1 – Е2. При условия постоянства потенциала электрода сравнения и определенной, заведомо известной величины его, по величине э.д.с. можно определить потенциал индикаторного электрода. Но измеряемое напряжение V k (клеммовое напряжение) будет меньше разницы электродных потенциалов (э.д.с.) на величину падения напряжение IR i:
 (4.1)
(4.1)
Где Rj – внутреннее сопротивление ячейки.
Точное измерение э.д.с. возможно лишь в том случае, когда I = 0, то есть в отсутствии тока. Этого можно добиться встречным включением напряжения, соответствующего э.д.с. (компенсационный метод Поггендорфа), или применением измерительных приборов с высоким входным сопротивлением.
В компенсационном методе на электроды ячейки налагают э.д.с. внешнего источника постоянного тока (батареи или аккумулятора) противоположно направленную э.д.с. гальванической ячейки. При установившейся компенсации, когда э.д.с. ячейки и источника равны, в цепи тока нет. Величину компенсирующей э.д.с. определяют, сравнивая ее с э.д.с. нормального элемента Вестона. На компенсационном методе основан принцип работы потенциометров, например, типа Р - 307 и др. Для измерений в неводных средах, со стеклянными и некоторыми другими электродами, имеющими высокое сопротивление, эти приборы малопригодны.
Другой способ заключается в использовании приборов с большим (до 1011 – 1015 Ом) входным (внешним) сопротивлением, благодаря чему измерения можно проводить практически без мешающего действия тока. Этот способ воплощен в рН–метрах, ионометрах, электронных вольтметрах. Эти приборы удобны в работе, так как измерения проводятся быстро и результаты измерений можно считывать со шкалы (в рН–метрах, ионометрах) или цифрового индикатора (в электронных вольтметрах) [7].
Электроды сравнения. Согласно электрохимическим реакциям, протекающим на поверхности раздела металл-раствор, различают электроды первого, второго и третьего рода.
К электродам первого рода относятся металлы, потенциалы которых обратимы относительно своих ионов, как, например, серебряный электрод в растворе содержащем катионы серебра.
Потенциалы электродов второго рода обратимы относительно своих анионов, образующих с катионами металла электрода малорастворимый осадок; например, серебряный электрод в насыщенном растворе хлорида серебра в присутствии анионов хлора.
Электроды третьего рода представляют собой металлы, которые находятся в равновесии с раствором, насыщенном двумя малорастворимыми электролитами с одним общим анионом и катионами, один из которых является ионом металла электрода, а второй посторонним, находящимся в избытке [6].
По международному соглашению в качестве стандартного электрода принят стандартный водородный электрод, потенциал которого при любой температуре считают равным нулю. Но водородный электрод малопригоден в обычных условиях работы и на практике используются специально изготовленные электроды сравнения, в основном это насыщенный каломельный и хлорсеребряный электроды.
Электрод сравнения должен иметь постоянный, воспроизводимый потенциал даже в условиях слабых токов. Их обычно изготавливают в виде компактных полуэлементов, чтобы можно было погружать непосредственно в анализируемый раствор; роль солевого мостика играет очень небольшое отверстие (точечное отверстие; узкая щель) во внешней оболочке. Потенциалы этих электродов при их изготовлении измеряют относительно стандартного водородного электрода и дают в характеристике (паспорте) электрода.
Насыщенный каломельный и хлорсеребряный электроды применяют как электроды сравнения практически во всех случаях, так как потенциалы их не зависят от состава изучаемого раствора [7].
Индикаторные электроды. Электрод, потенциал которого зависит от концентрации определяемого иона в растворе, называется индикаторным (измерительным).
Зависимость величины электродного потенциала от концентрации ионов в растворе, к которым чувствителен электрод, в идеальном случае выражается уравнением Нернста:
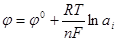 (4.2)
(4.2)
где φ0 - стандартный электродный потенциал; R - газовая постоянная; Т - абсолютная температура; n - заряд иона; F - число Фарадея; а i - активность иона.
В разбавленных растворах коэффициент активности близок к единице, поэтому активность можно заменить концентрацией:
 (4.3)
(4.3)
При 25°C и соответствующих значениях R, F, переходя от натуральных логарифмов к десятичным, получим:
 (4.4)
(4.4)
Одним из основных требований к индикаторным электродам, кроме определенной зависимости от активности (концентрации), является воспроизводимость и быстрота отклика потенциала при изменении активности иона.
Известны два основных вида индикаторных электродов – металлические и мембранные.
Металлические индикаторные электроды можно изготавливать из различных металлов, способных давать обратимые полуреакции, например, из серебра, меди, ртути, свинца, кадмия. Потенциалы этих металлов воспроизводимо и предсказуемо отражают активность их ионов в растворе. Металлические электроды служат не только для определения собственных ионов; косвенно они чувствительны к анионам, образующим малорастворимые осадки с этими ионами. В этом случае необходимо только насытить изучаемый раствор малорастворимой солью. Например, серебряный электрод, чувствительный к ионам серебра, будет правильно отражать и концентрацию хлорид ионов в растворе, насыщенном хлоридом серебра. И, если по отношению к ионам Ag+ он будет электродом первого рода, то при определении Сl- - электродом второго рода, поскольку он измеряет концентрацию ионов, не участвующих непосредственно в процессе переноса электронов, то есть в электродной реакции.
В окислительно-восстановительных реакциях в качестве индикаторных электродов применяют инертные металлы (платину, золото), потенциал которых зависит от отношения концентраций окисленной и восстановленной форм одного или нескольких веществ в растворе. Металлические электроды изготавливают из проволоки или пластинки. Перед работой поверхность их тщательно очищают. Хорошим способом очистки для многих электродов является быстрое погружение их в концентрированную азотную кислоту и последующее многократное промывание дистиллированной водой.
В отличие от рассмотренных электродов, потенциал которых определяется процессами переноса электронов между ионами в растворе и металлом (то есть электрохимической реакцией), в мембранных ионоселективных электродах возникновение потенциала связано с процессом обмена ионов между мембраной и раствором.
Процесс переноса через мембрану состоит из двух фаз: проникновение иона в мембрану и перемещение иона внутри мембраны. При этом на поверхности возникает потенциал, который начинает препятствовать дальнейшему перемещению ионов и, в конечном счете, устанавливается динамическое равновесие, при котором устанавливается потенциал, подчиняющийся уравнению Нернста. Мембранный электрод – это устройство цилиндрической трубчатой формы, в котором ионоселективная мембрана разделяет внутренний и внешний (анализируемый) растворы и одновременно служит средством электролитического контакта между ними.
Во внутренний раствор постоянного состава, содержащий ионы, к которым селективна мембрана, опущен токоотводящий электрод, обеспечивающий контакт мембранного электрода с измерительным прибором. Причем для токоотводящего электрода (обычно это платиновая или серебряная проволока) созданы условия постоянства его потенциала.
Существующие ионоселективные электроды можно разделить на электроды с твердой мембраной, жидкой мембраной и стеклянные электроды.
Активный компонент твердой мембраны - малорастворимое соединение (в виде моно- или поликристаллов, осадков, твердых ионообменников и др.), обладающие ионной проводимостью. Электроды с такой мембраной "откликающиеся" на одноименные ионы (один из ионов, входящих в состав самого компонента) и часто еще на противоионы связанные с первыми величиной ПР.
Жидкостная мембрана представляет собой не смешивающуюся с водой органическую жидкость (растворитель с растворенным в нем ионообменным веществом), которая обладает селективным свойством проникновения через нее определенных ионов. Органическая и водная фазы в электроде отделены друг от друга полупроницаемой, инертной мембраной. Известны катионо- и анионоселективные жидкие мембраны. Основное отличие жидких мембран от твердых заключается в том, что они содержат подвижные ионогенные группы.
Стеклянные электроды, хотя и имеют твердую мембрану из ионоселективного стекла, по механизму ближе (аналогичны) электродам с жидкой мембраной.
Широкое применение получили рН – чувствительные стеклянные электроды. Кроме того, разработаны различные сорта ионоселективных стекол, способных к обмену с соответствующими катионами (в основном однозарядными), из которых изготавливают катиончувствительные электроды.
Потенциометрические методы анализа можно разделить на прямую потенциометрию (ионометрию) и потенциометрическое титрование.
Прямая потенциометрия. Уравнение Нернста даст простое соотношение между потенциалом электрода и активностью соответствующего иона в растворе. Поэтому на основания измеренной э.д.с. гальванического элемента, зная потенциал электрода сравнения, можно вычислить потенциал индикаторного электрода, а затем рассчитать активность и концентрацию определяемого иона. Однако здесь возникают некоторые трудности.
Во-первых, реально измеряемая э.д.с. гальванического элемента включает в себя кроме потенциалов электродов и диффузионный потенциал (потенциал жидкостного соединения), который возникает между анализируемым раствором и внешним раствором электрода сравнения. Этот потенциал может достигать десятков милливольт, но точно измерить или оценить теоретичёски его невозможно. Его можно свести к минимуму, используя для соединения между растворами солевой мостик, состоящий из концентрированного раствора электролита, ионы которого имеют одинаковую подвижность, например, насыщенного раствора хлорида калия. При этом потенциал жидкостного соединения составляет обычно несколько милливольт или меньше, то есть имеет несущественную для большинства электроаналитических методов величину, за исключением прямой потенциометрии, так как изменение потенциала (или погрешность в измеряемой э.д.с.) даже на 1 мВ дает относительную ошибку до 4 %.
Во-вторых, для вычисления активности определяемого иона по уравнению Нернста надо знать величину стандартного потенциала индикаторного электрода, однако для многих электродов в первую очередь это касается мембранных (которые в основном и используются как индикаторные в ионометрии), это неизвестный потенциал ассиметрии, величина которого изменяется во времени.
В-третьих, из уравнения Нернста можно определить активность, а для вычисления концентрации определяемого вещества требуется знание коэффициентов активности, которые, как правило, недоступны, поскольку обычно состав, а значит и ионная сила раствора, неизвестны. Поэтому при использовании прямой потенциометрии для аналитических целей применяют, обычно, эмпирическую калибровку измерительного электрода.
К достоинствам прямой потенциометрии следует отнести возможность анализа чрезвычайно малых проб и отсутствие изменения или разложения пробы.
Очень ценным качеством прямой потенциометрии (это особенно касается метода калибровки) является возможность автоматизации измерений, а следовательно, и автоматизации контроля производства, уровня загрязнения в производстве, сточных вод и т.д.
Еще более широкое применение в аналитической химии нашло потенциометрическое титрование, которое заключается в потенциометрическом наблюдении за ходом химической реакции. Потенциометрическое титрование можно охарактеризовать как титрование, при котором изменение э.д.с. гальванического элемента записывают в виде функции добавленного титранта.
Для этого необходимо только, чтобы в реакции участвовал ион, для которого существует подходящий индикаторный электрод.
Установка для проведения потенциометрического титрования проста. Она включает в себя сосуд для титрования (химический стакан), в который помещают анализируемый (титруемый) раствор, подходящие электроды – индикаторный и сравнения, опущенные в этот раствор, приспособление для эффективного перемешивания титруемого раствора (обычно, это магнитная мешалка), бюретку со стандартным раствором титранта и измерительный прибор.
Титрант в начале титрования можно добавлять довольно быстро. При приближении к моменту эквивалентности его следует вводить малыми порциями и ждать установления равновесия перед добавлением новой порции, так как необходимо, время для приобретения индикаторным электродом устойчивого потенциала. В противоположность прямой потенциометрии при титровании измерение э.д.с. с высокой точностью не сложно, так как изменение потенциала индикаторного электрода вблизи точки эквивалентности довольно большое.
Главная цель этого метода – установление с высокой точностью точки эквивалентности, то есть объема титранта, точно известной концентрации, пошедшего на взаимодействие с определяемым веществом. Зная эквивалентный объем титранта, рассчитывают количество определяемого вещества по формуле:
 (4.5)
(4.5)
где VT - эквивалентный объем титранта, мл; NT - концентрация титранта, н; Э - эквивалент определяемого вещества.
Для определения точки эквивалентности применяют различные инструментальные, графические и расчетные методы. Выбор метода зависит от удобства его применения, характера кривой титрования и допустимой погрешности определения.
Один из простых и удобных методов определения точки эквивалентности, который чаще всего и применяется - нахождение ее по кривой титрования, построенной в координатах: э.д.с. - объем титранта. Причем значения э.д.с. могут быть выражены как в единицах напряжения (мВ, В), так и в других условных единицах рН, деления шкалы - L и др.). Точку эквивалентности определяют, как обычно, на середине скачка титрования. Если скачок не ярко выражен, что наблюдается, например, при титровании разбавленных растворов, то более точно точку эквивалентности находят по дифференциальной кривой титрования, построенной в координатах ∆Е/∆V - V (∆рН/∆V - V, ∆L/∆V - V), которая в точке эквивалентности дает резкий максимум.
Если э.д.с, соответствующую точке эквивалентности, установить предварительно из подобных титрований или из теоретических расчетов, можно просто титровать до тех пор, пока э.д.с. не приобретет необходимое значение. Этот принцип заложен в основу автоматического потенциометрического титрования.
Потенциометрическое титрование по своим возможностям значительно превосходит титрование с цветными химическими индикаторами, обладая целым рядом преимуществ:
1. более высокой точностью определения, так как субъективная оценка конца титрования заменяется объективными показателями чувствительных приборов;
2. большой чувствительностью, позволяющей определять очень малые количества веществ;
3. возможностью титровать мутные иди окрашенные растворы, исключающие использование индикаторов;
4. возможностью дифференцированно и последовательно определить два и более компонентов в данной порции исследуемого раствора;
5. возможностью автоматизировать титрование, что позволяет уменьшить трудоемкость метода;
6. возможностью получить и термодинамическую информацию о константах диссоциации слабых электролитов, константах образования комплексных ионов.
В потенциометрическом титровании нет необходимости знать потенциал электрода сравнения, стандартные потенциалы индикаторных электродов и значения жидкостных диффузионных потенциалов. В то же время потенциометрическое титрование позволяет:
1. использовать для определения большее число индикаторных электродов, так как во многих случаях требования к электродам в отношении постоянства угла наклона электродной функции и стандартного потенциала менее жесткие, потому что важно не абсолютное значение потенциала (и э.д.с. элемента), а его изменение, связанное с добавленным титрантом;
2. вести измерение в присутствии мешающих веществ, влияющих на потенциал индикаторного электрода, путем подбора титранта, селективно реагирующего только с определяемым веществом.
Широкие возможности объясняются тем, что в потенциометрическом титровании могут быть использованы все четыре типа реакций: кислотно-основные, осаждения, комплексообразования и окисления-восстановления.
Выбор индикаторного электрода при потенциометрическом титровании определяется типом протекающей реакции, либо природой определяемых ионов или ионов титранта, участвующих в реакции, а также удобством работы с электродами.
При окислительно-восстановительном титровании в качестве индикаторных используют электрод из платины или другого благородного металла, при кислотно-основном титровании изменение концентрации ионов водорода измеряют с помощью рН - чувствительных электродов, чаще всего стеклянных, в реакциях осаждения и комплексообразования выбирают электрод, чувствительный к определяемому иону или реагирующему с ним.
Для того чтобы провести анализ потенциометрическим титрованием, необходимо:
1. анализируемый объект (пробу) перевести в раствор;
2. выбрать соответствующий титрант, то есть подобрать химическую реакцию для определяемого вещества, отвечающую требованиям, предъявленным к реакциям в титриметрии;
3. создать условия для проведения данной реакции (рН, t, отсутствие мешающих определению компонентов);
4. подобрать правильно электроды. Так как в качестве электродов сравнения обычно используются каломельный или хлорсеребряный электроды, то задача сводится к правильному выбору индикаторного электрода;
5. перенести анализируемый раствор или его определенную долю (аликвотную часть) в сосуд для титрования и опустить туда электроды, подсоединив их к измерительному прибору;
6. подвести бюретку с титрантом к сосуду для титрования и титровать при эффективном перемешивании, записывая результаты измерений объема добавляемого титранта по бюретке и э.д.с. (рН или L) по шкале измерительного прибора;
7. по данным измерений построить графическую зависимость э.д.с. (pH, L) от объема прибавляемого титранта (кривую титрования), определить точку эквивалентности, то есть эквивалентный объем титранта, пошедшего на взаимодействие с определяемым веществом, и рассчитать его количество в граммах по формуле (4.5).
Осадительное потенциометрическое титрование. Осадительное титрование основано на реакции осаждения определяемого вещества (иона) в процессе титрования. При подборе реакции осаждения (титранта) надо руководствоваться требованиями к титриметрическим реакциям и законом, управляющим процессом осаждения, - правилом произведения растворимости. Необходимо создать все условия, чтобы концентрация определяемого (осаждаемого) иона, оставшегося в растворе после осаждения, не превышала 10-6 моль/л.
Основным параметром, позволяющим контролировать ход реакции, является концентрация осаждаемого иона или иона осадителя. Потенциометрический контроль реакции, то есть потенциометрическое титрование возможно, если есть возможность подобрать соответствующий индикаторный электрод.
В осадительном титровании индикаторными электродами могут быть мембранные электроды, чувствительные к одному из ионов, участвующих в реакции осаждения, или металлические электроды, чувствительные к собственному (одноименному) иону, если этот ион осаждается или осаждает: серебряный электрод для определения Ag( I), ртутный - для определения Hg(II), медный - для определения Cu(II) и др. Например, серебряный электрод (серебряная проволока или пластина) может служить индикаторным при определении серебра, а также некоторых ионов (С l-, В r-, I-, CN-, CNS- и др.), образующих малорастворимые соли серебра при титровании их раствором азотнокислого серебра.
Электродом сравнения могут служить каломельный и хлорсеребряный электроды. Если в реакции осаждения участвуют ионы хлора, то для предотвращения диффузии хлорида калия в пробу и уменьшения погрешности электрод сравнения можно поместить в отдельный раствор, соединив его с титруемым солевым мостиком из нитрата калия [7].
Определение содержания галогенидов в растворе. Основной задачей является определение хлоридов и иодидов в растворе при совместном присутствии. Для решения ее проводятся два потенциометрических титрования.
1) титрование раствора AgNO3 стандартным раствором NaCl для определения концентрации, то есть для стандартизации раствора AgNO3;
2) титрование анализируемого раствора (С l- и I-) раствором AgNO3, концентрация которого установлена первым титрованием.
В основе первого титрования лежит реакция:
Ag+ + C l- = AgCl
При титровании смеси иодидов и хлоридов:
I- + Ag+ = AgI
С l- + Ag+ = AgCl
Значительная разница ПР (  ) позволяет сделать выводы:
) позволяет сделать выводы:
1) вначале титруется I- с образованием менее растворимого AgI, а затем – С l-;
2) С l- начинает осаждаться только тогда, когда I- будет полностью осажден (оттитрован), и концентрация Ag+ достигнет величины, необходимой для достижения и превышения ПР AgCl: 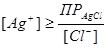 ;
;
3) получится два четко раздельных скачка на кривой титрования, соответствующие: первый – оттитровыванию I-, второй – С l- (рис. 4.3).
В работе используется серебряный электрод как индикаторный, так как потенциал его определяется концентрацией ионов серебра:
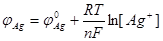 (4.6)
(4.6)
 (4.7)
(4.7)
где φ0 Ag = 0,8 В, [ Ag+] - концентрация ионов серебра в данный момент титрования, г-ион/л.
При титровании раствора AgNO3 раствором NaCl концентрация ионов серебра уменьшается, и потенциал серебряного электрода будет соответственно уменьшаться. Можно теоретически рассчитать изменение потенциала серебряного электрода (а также э.д.с. гальванического элемента) в процессе титрования, и экспериментально это подтвердить потенциометрическими измерениями.
До прибавления эквивалентного объема NaCl, т.е. до точки эквивалентности потенциал серебряного электрода рассчитывается по уравнению (4.7), где [ Ag+] – концентрация ионов серебра, еще не вступивших в реакцию с хлоридом; в момент эквивалентности, при прибавлении эквивалентного количества осадителя  и поэтому
и поэтому
 (4.8)
(4.8)
после точки эквивалентности при избытке осадителя  , где [ Cl-] определяется избытком титранта
, где [ Cl-] определяется избытком титранта
 (4.9)
(4.9)
ПР - const, поэтому
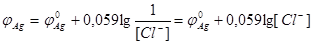 (4.10)
(4.10)
Кривая титрования будет иметь вид, изображенный на рис. 4.1.

Рис. 4.1. Кривая титрования AgNO3 раствором NaCl.
При титровании галогенида, например С l-, раствором AgNO3 потенциал серебряного электрода будет увеличиваться, т.к. концентрация ионов серебра возрастает. До точки эквивалентности, когда переведены в осадок еще не все ионы хлора , где [ Cl-] - концентрация непрореагировавших ионов, г-ион/л.
Соответственно потенциал индикаторного электрода находят по уравнению (4.10). В точке эквивалентности, когда ионы хлора практически будут осаждены, а концентрация ионов серебра станет равной концентрации ионов хлора в растворе 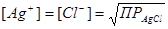 , потенциал индикаторного электрода по уравнению (4.8). После точки эквивалентности, концентрация ионов серебра будет определяться избытком титранта и φ Ag рассчитывают по (4.7). Кривая титрования показана на рис. 4.2.
, потенциал индикаторного электрода по уравнению (4.8). После точки эквивалентности, концентрация ионов серебра будет определяться избытком титранта и φ Ag рассчитывают по (4.7). Кривая титрования показана на рис. 4.2.

Рис. 4.2. Кривая титрования хлоридов раствором AgNO3.
При потенциометрическом титровании смеси иодидов и хлоридов получится кривая титрования (рис. 4.3) с двумя скачками, соответствующими моментам эквивалентности реакций осаждения иодидов и хлоридов [7].

Рис 4.3. Кривая титрования иодидов и хлоридов раствором AgNO3.
Методика проведения анализа. Дифференцированное определение I- и Cl- в их смеси проводят титрованием ~ 0,05 н. стандартным раствором нитрата серебра с серебряным индикаторным электродом и Нас.КЭ (насыщенным каломельным электродом) сравнения. Э.д.с. потенциометрической ячейки измеряют компенсационным методом.
Пока в растворе присутствуют ионы иодида, потенциал Нас.КЭ больше потенциала серебряного электрода, поэтому последний должен быть подключен к отрицательной клемме потенциометра [6].
До начала работы необходимо:
1) подключить измерительный прибор к электросети для прогревания в течение 15-20 минут;
2) зачистить наждачной бумагой металлическую часть индикаторного (серебряного) электрода и промыть дистиллированной водой весь электрод;
3) протереть фильтровальной бумагой и промыть дистиллированной водой электрод сравнения;
4) тщательно протереть фильтровальной бумагой и промыть дистиллированной водой магнитный элемент.
Работа состоит из двух частей:
а) Определение методом потенциометрического титрования концентрации раствора AgN O3 (  ).
).
В чистый стакан для титрования отобрать с помощью бюретки 3 мл раствора AgN O3 поместить в стакан магнитный элемент и опустить электроды так, чтобы они не касались магнитного элемента в покое и в дальнейшем при перемешивании раствора. Прилить в стакан дистиллированной воды столько, чтобы рабочие части электродов (металлическая часть индикаторного и место контакта с раствором электрода сравнения) были полностью погружены в раствор. Включить магнитную мешалку и подобрать подходящий режим перемешивания. Включить измерительный прибор и измерить начальную величину э.д.с. полученного гальванического элемента. Затем титровать стандартным раствором NaCl, прибавляя его по 0,2 мл и измеряя э.д.с. после каждой порции прибавленного титранта. Полученные измерения заносят в таблицу. В начале титрования э.д.с. мало изменяется, затем вблизи точки, эквивалентности происходит резкое, скачкообразное изменение, после конца скачка опять наблюдаются близкие по значению величины э.д.с. Титровать необходимо до получения 6 - 7 измерений близких по значению величии э.д.с. после конца скачка титрования (~ до 140 - 150 мВ, т.е. 0,14 -0,15 В).
б) Определение методом потенциометрического титрования содержания хлоридов и иодидов при совместном присутствии.
Полученный у лаборанта анализируемый раствор, содержащий смесь солей K I и NaCl, титруют раствором AgN O3 по выше описанной методике. Измерения записывают в таблицу. В отличие от предыдущего титрования здесь должно получиться два скачка титрования, то есть два резких скачкообразных изменения э.д.с. Титрование заканчивают, добавив 6 - 7 порций титранта после конца второго скачка титрования (~ до 420 - 450 мВ, т.е. до 0,42-0,45 В).
По полученным данным строят кривые титрования - графические зависимости э.д.с. от объема (V) титранта (рис. 4.1, 4.2, 4.3). На скачках титрования находят точки эквивалентности и соответствующие им эквивалентные объемы титрантов, израсходованные на взаимодействие с титруемыми веществами.
Рассчитывают:
Концентрацию AgN O3 по формуле:
 (4.11)
(4.11)
где 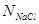 - концентрация стандартного раствора NaCl, взятого для титрования, н;
- концентрация стандартного раствора NaCl, взятого для титрования, н;  - объем стандартного раствора NaCl, пошедший на титрование AgNO3, мл. (Рис. 4.1);
- объем стандартного раствора NaCl, пошедший на титрование AgNO3, мл. (Рис. 4.1);
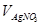 - объем раствора AgNO3, отобранный для титрования, 3,00 мл.
- объем раствора AgNO3, отобранный для титрования, 3,00 мл.
Содержание хлор-иона и иона йода в анализируемом растворе рассчитывают по формулам:
 (4.12)
(4.12)
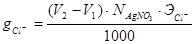 (4.13)
(4.13)
где V1 – объем AgNO3 пошедший на титрование иодида, соответствующий первому скачку на кривой титрования смеси солей, мл (рис 4.3); V2 – общий объем AgN O3, пошедший на титрование йодида и хлорида, соответствующий второму скачку на кривой титрования смеси солей, мл (рис. 4.3);  - концентрация раствора AgN O3, рассчитанная по формуле (4.11); Э Cl-, Э I- - эквиваленты хлора и йода, равные их атомным массам.
- концентрация раствора AgN O3, рассчитанная по формуле (4.11); Э Cl-, Э I- - эквиваленты хлора и йода, равные их атомным массам.
Содержание KI и KCl а анализируемом растворе рассчитывается но формулам:
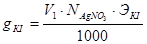 (4.14)
(4.14)
 (4.15)
(4.15)
где Э KI и Э KCl - эквиваленты KI и KCl, равные их молекулярным массам [7].
|
из
5.00
|
Обсуждение в статье: Выбор метода анализа ключевого компонента. |
|
Обсуждений еще не было, будьте первым... ↓↓↓ |

Почему 1285321 студент выбрали МегаОбучалку...
Система поиска информации
Мобильная версия сайта
Удобная навигация
Нет шокирующей рекламы