 |
Главная |


Классификация методов комплексонометрического титрования
|
из
5.00
|
- Метод прямого титрования. Анализируемый раствор, содержащий катионы определяемого металла, разбавляют в мерной колбе. Берут аликвотную часть для титрования. Предварительно титруемый раствор доводят до определенного значения рН при помощи буферных растворов. Наряду с буферным раствором иногда добавляют еще вспомогательный агент (тартрат, цитрат и др.), связывающий некоторые катионы и не дающий им возможность выпадать в виде гидроксидов в щелочном растворе.
Титрование ведут стандартным раствором ЭДТА в щелочной среде ( аммиачный буфер NH3 + NH4Cl с pH = 8-9) с индикатором – эриохром черный Т или в кислой среде с ксиленоловым оранжевым.
В процессе прямого титрования концентрация определяемого катиона сначала постепенно снижается, затем вблизи точки эквивалентности резко падает. Этот момент отмечают по изменению окраски индикатора, мгновенно реагирующего на изменение концентрации катионов металла-комплексообразователя.
- Метод обратного титрования
Используют в случае, когда:
- невозможно провести прямое титрование;
- нет подходящего индикатора на катионы определяемого металла;
- в буферном растворе катионы образуют осадки;
- реакция комплексообразования протекает медленно;
- нужно определить содержание Ca2+, Mg2+ в нерастворимых в воде осадках.
К анализируемому раствору прибавляют точно измеренный объем стандартного раствора комплексона, нагревают до кипения для завершения реакции комплексообразования,и затем на хододе оттитровывают избыток комплексона титрованным раствором сульфата магния или сульфата цинка.
Для установления точки эквивалентности применяют металл-индикатор, реагирующий на ионы Mg2+ или Zn2+..
Условия проведения комплексонометрического титрования.
- В точке эквивалентности определяемые катионы должны быть практически полностью связаны в комплекс. Константа нестойкости его должна быть незначительной ( Кнестойк катион-комплексон).
- Определяемые катионы должны образовывать с металл-индикатором комплексы менее прочные, чем комплексы катионов с комплексоном.
- рН титруемого раствора поддерживается с помощью буферов: щелочной - NH3 + NH4Cl с pH = 8-9, кислый – 0,1н.HCl.
Определение содержания Ca и Mg в растворе. Индикаторы комплексонометрии являются органическими красителями. Они образуют с ионами металлов окрашенные комплексные соединения - - металл-индикаторы, которые являются менее прочными, чем комплекс этого же металла с титрантом-комплексоном.
Метод основан на образовании прочных, растворимых в воде комплексов кальция и магния с трилоном Б при определенном значении pH среды в присутствии индикатора кислотного хром темно-синего.
Определению содержания кальция и магния мешают железо, медь и цинк, которые связывают в комплекс диэтилдитиокарбаминатом натрия (ДДК), а затем из одной пробы при разном значении pH среды определяют отдельно содержание кальция и магния титрованием трилоном Б с индикатором кислотным хром темно-синим.
Расчет содержания кальция и магния выполняют по формулам:
a х N х 20
С = ----------,
Ca V
b х N х 12,6
С = ------------,
Mg V
где:
a и b - расход раствора трилона Б (титранта), пошедшего на титрование ионов кальция и магния, куб. см;
N - нормальность раствора трилона Б;
V - объем пробы, взятой для анализа, куб. см;
20 и 12,16 - грамм-эквиваленты кальция и магния.
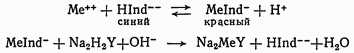
Особенности протекания реакции, благодаря которым комплексонометрия получила широкое применение в химическом анализе:
- в результате реакции образуются комплексы только одного состава с соотношением металл:лиганд равным 1:1 (комплексонаты); при этом комплексонаты бесцветны, хорошо растворимы в воде и обладают высокой устойчивостью, так как центральный атом металла прочно связан полидентатным хелатным лигандом;
- реакция является обратимым процессом и может быть сдвинута как в сторону образования, так и в сторону разрушения комплексоната, что легко достигается с помощью варьирования величины рН раствора – подкисление приводит к смещению равновесия влево к исходным реагентам, а подщелачивание способствует образованию комплексоната;
- в результате реакции выделяются ионы водорода, поэтому её следует проводить в буферной среде, поддерживая оптимальное значение рН, определяемое константой устойчивости комплексоната.
Условия комплексонометрического титрования. Основным условием комплексонометрического титрования является требование, предъявляемое к реакции, которая должна протекать таким образом, чтобы в точке эквивалентности определяемые катионы были практически полностью связаны в комплекс. Константа нестойкости таких комплексов должна быть очень незначительной. При этом определяемые катионы должны образовывать с металл-индикатором комплексы, отличающиеся меньшей прочностью, чем их комплексы с комплексоном.
Титрование комплексоном III проводится при строго определенных условиях, из которых наибольшее значение имеет соблюдение требуемого значения pH титруемого раствора.
В сильно кислых растворах образуются менее устойчивые кислые комплексные соединения. Комплексообразованию устойчивых комплексных соединений способствует повышение значения pH титруемого раствора. Следует также иметь в виду, что при образовании комплекса определяемого катиона с комплексоном освобождаются ионы водорода, pH раствора понижается. Поэтому, если титруемые растворы не защищены действием буферной смеси, понижение pH раствора может достигнуть нескольких единиц и требуемые комплексные соединения не образуются. Чтобы поддержать pH раствора на заданном уровне, необходимо проводить титрование в буферных растворах, отвечающих определенному значению pH.
Определение ОЖВ.Различают устранимую (временную) жесткость воды и постоянную. Первая определяется наличием в воде бикарбонатов магния и кальция (при кипячении они выпадают в виде соответствующих карбонатов), вторая - наличием сульфатов магния и кальция. Суммарное количество кальциевых и магниевых солей в 1 литре воды определяет общую жесткость.
Для определения общей жесткости анализируемую воду подщелачивают аммиачной буферной смесью до рН = 9. В качестве индикатора используют эриохром черный Т. В точке эквивалентности винно-красная окраска раствора меняется на синюю вследствие накопления анионов индикатора HInd2-.
Методика определения. Пипеткой емкостью 50,0 или 100,0 см3 отбирают пробу анализируемой воды, добавляют 5 см3 аммиачной буферной смеси, добавляют 20-30 мг индикатора - эриохрома черного Т и титруют раствором комплексона III до перехода окраски из винно-красной в синюю.
Жесткость воды - суммарное количество миллимолей кальция и магния, содержащееся в 1 л воды, рассчитывают по формуле:
Индикаторы. Кислотный хром темно-синий, кислотный хромоген черный специальный (эриохром черный Т) – в щелочной среде синие. Mg2, Са2+ + ЭДТА →бесцветный (Mg2, Са2+ + индикатор) + ЭДТА---------< Вишнево-красный цвет индикатор→синий
|
*37)Оптические методы анализа. Фотометрия. Закон Бугера-Ламберта-Бера. ОПТИЧЕСКИЕ МЕТОДЫ АНАЛИЗА. К оптическим методам анализа относят физико-химические методы, основанные на взаимодействии электромагнитного излучения с веществом. Это взаимодействие приводит к различным энергетическим переходам, которые регистрируются экспериментально в виде поглощения излучения, отражения и рассеяния электромагнитного излучения. Оптические методы включают в себя большую группу спектральных методов анализа. В методах атомной спектроскопии мы имеем дело с узкими линейчатыми спектрами, а в методах молекулярной спектроскопии – с широкими слабоструктурированными спектрами. Это определяет возможность их применения в количественном анализе и требования, предъявляемые к измерительной аппаратуре – спектральным приборам. 1.1. Фотометрический метод анализа. 1.1.1. Основные законы и формулы: Фотометрический анализ относится к абсорбционным методам, т.е. основан на измерении поглощения света веществом. Он включает спектрофотометрию, фотоколориметрию и визуальную фотометрию, которую обычно называют колориметрией. Каждое вещество поглощает излучение с определенными (характерные только для него) длинами волн, т.е. длина волны поглощаемого излучения индивидуальна для каждого вещества, и на этом основан качественный анализ по светопоглошению.Основой количественного анализа является закон Бугера-Ламберта-Бера: А = e l c, где А = –lg (I / I0) = –lg T – оптическая плотность; I0 и I – интенсивность потока света, направленного на поглощающий раствор и прошедшего через него;с – концентрация вещества, моль/л; l – толщина светопоглощающего слоя;e - молярный коэффициент светопоглощения; T - коэффициент пропускания. Для определения концентрации анализируемого вещества наиболее часто используют следующие методы: 1) молярного коэффициента светопоглощения; 2) градуировочного графика; 3) добавок; 4) дифференциальной фотометрии; 5) фотометрического титрования. Метод молярного коэффициента поглощения. При работе по этому методу определяют оптическую плотность нескольких стандартных растворов Аст, для каждого раствора рассчитывают e = Аст / (lсст) и полученное значение e усредняют. Затем измеряют оптическую плотность анализируемого раствора Ах и рассчитывают концентрацию сх по формуле сх = Ах /(el). Ограничением метода является обязательное подчинение анализируемой системы закону Бугера-Ламберта-Бера, по крайней мере, в области исследуемых концентраций. Метод градуировочного графика. Готовят серию разведений стандартного раствора, измеряют их поглощение, строят график в координатах Аст – Сст. Затем измеряют поглощение анализируемого раствора и по графику определяют его концентрацию. Метод добавок. Этот метод применяют при анализе растворов сложного состава, так как он позволяет автоматически учесть влияние «третьих» компонентов. Сущность его заключается в следующем. Сначала определяют оптическую плотность Ах анализируемого раствора, содержащего определяемый компонент неизвестной концентрации сх, а затем в анализируемый раствор добавляют известное количество определяемого компонента (сст) и вновь измеряют оптическую плотность Ах+ст. Оптическая плотность Ах анализируемого раствора равна Ах = e l cх, а оптическая плотность анализируемого раствора с добавкой стандартного Ах+ст = e l (cх + сст). Концентрацию анализируемого раствора находим по формуле: сх = сст Ах / (Ах+ст – Ах). Метод дифференциальной фотометрии. Если в обычной фотометрии сравнивается интенсивность света, прошедшего через анализируемый раствор неизвестной концентрации, с интенсивностью света, прошедшего через растворитель, то в дифференциальной фотометрии второй луч света проходит не через растворитель, а через окрашенный раствор известной концентрации – так называемый раствор сравнения. Фотометрическим методом можно определять также компоненты смеси двух и более веществ. Эти определения основаны на свойстве аддитивности оптической плотности: Асм = А1 + А2 + …+ Аn, где Асм - оптическая плотность смеси; А1 , А2, Аn – оптические плотности для различных компонентов смеси. Фотометрические методы анализа применяются для контроля разнообразных производственных процессов. Эти методы могут быть применены для анализа больших и малых содержаний, но особенно ценной их особенностью является возможность определения примесей (до 10-5...10-6%). Методы абсорбционной спектроскопии используют в химической, металлургической, фармацевтической и других отраслях, а также в медицине и сельскохозяйственном производстве. Промышленностью выпускаются приборы для абсорбционной спектроскопии: колориметры, фотометры, фотоэлектроколориметры, спектрофотометры и т. д., в которых используют различные комбинации осветителей, монохроматоров и приемников света. *38)Обзор электрохимических методов анализа. ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА.Электрохимические методы анализа и исследования основаны на изучении и использовании процессов, протекающих на поверхности электрода или в приэлектродном пространстве. Любой электрический параметр (потенциал, сила тока, сопротивление и др.), функционально связанный с концентрацией анализируемого раствора и поддающийся правильному измерению, может служить аналитическим сигналом. Различают прямые и косвенные электрохимические методы. В прямых методах используют зависимость силы тока (потенциала и т.д.) от концентрации определяемого компонента. В косвенных методах силу тока (потенциал и т. д.) измеряют с целью нахождения конечной точки титрования определяемого компонента подходящим титрантом, т.е. используют зависимость измеряемого параметра от объема титранта. Для любого рода электрохимических измерений необходима электрохимическая цепь или электрохимическая ячейка, составной частью которой является анализируемый раствор. 2.1. Потенциометрический метод анализа.2.1.1. Основные законы и формулы: Потенциометрические методы основаны на измерении разности потенциалов индикаторного электрода и электрода сравнения или, точнее, электродвижущих сил (ЭДС) различных цепей, поскольку экспериментально измеряется именно ЭДС, являющаяся разностью потенциалов. Равновесный потенциал индикаторного электрода связан с активностью и концентрацией веществ, участвующих в электродном процессе, уравнением Нернста: Е = Е° + R T/(n F) ln (аокис/авосст), Е = Е° + R T /(n F) ln ([окисл] үокисл /([восст] үвосст)), R - универсальная газовая постоянная, равная 8,31 Дж/(моль . К); Т - абсолютная температура; F- постоянная Фарадея (96500 Кл/моль); n - число электронов, принимающих участие в электродной реакции; аокис, авосст - активности соответственно окисленной и восстановленной форм редокс-системы; [окисл] и [восст] - их молярные концентрации; үокис, үвосст - коэффициенты активности; Е° - стандартный потенциал редокс-системы.Подставляя Т = 298,15 К и числовые значения констант в уравнение, получаем:Е = Е° + (0,059 / n) lg (аокис/авосст), Е = Е° + (0,059 / n) lg ([окисл] үокисл/([восст] үвосст)). Методы прямой потенциометрии основаны на применении уравнения Нернста для нахождения активности или концентрации участника электродной реакции по экспериментально измеренной ЭДС цепи или потенциалу электрода. Наибольшее распространение среди прямых потенциометрических методов получил метод определения рН, но создание в последнее время надежно работающих ионоселективных электродов значительно расширило практические возможности прямых методов. Показатель рН измеряют и методом потенциометрического титрования. Для определения рН чаще всего используют стеклянный электрод. Основными достоинствами стеклянного электрода являются простота работы, быстрое установление равновесия и возможность определения рН в окислительно-восстановительных системах. К недостаткам относятся хрупкость материала электрода и сложность работы при переходе к сильнощелочным и сильнокислым растворам. Кроме концентрации ионов водорода, прямым потенциометрическим методом с ионоселективными электродами можно определить содержание нескольких десятков различных ионов. Потенциометрическое титрование основано на определении точки эквивалентности по результатам потенциометрических измерений. Вблизи точки эквивалетности происходит резкое изменение (скачок) потенциала индикаторного электрода. Так же, как и в других титриметрических методах, реакции потенциометрического титрования должны протекать строго стехиометрически, иметь высокую скорость и идти до конца. Для потенциометрического титрования собирают цепь из индикаторного электрода в анализируемом растворе и электрода сравнения. В качестве электродов сравнения чаще всего используют каломельный или хлорсеребряный электроды. Тип применяемого индикаторного электрода при потенциометрическом титровании зависит от свойств титриметрической смеси и ее взаимодействия с электродом. В кислотно-основном титровании используют стеклянный электрод, в окислительно-восстановительном - инертный (платиновый) электрод или электрод, обратимый по отношению к одному из ионов, содержащихся в титриметриметрической смеси; в осадительном – серебряный электрод; в комплексонометрическом - металлический электрод, обратимый к титруемому иону металла. Для нахождения точки эквивалентности часто строят дифференциальную кривую в координатах DЕ/DV – V. На точку эквивалентности указывает максимум полученной кривой, а отсчет по оси абсцисс, соответствующий этому максимуму, дает объем титранта, израсходованного на титрование до точки эквивалентности. Определение точки эквивалентности до дифференциальной кривой значительно точнее, чем по простой зависимости Е – V. Основными достоинствами метода потенциометрического титрования являются высокая точность и возможность проводить определения в разбавленных растворах, в мутных и окрашенных средах, а также определять несколько веществ в одном растворе без предварительного разделения. Значительно расширяется область практического применения потенциометрического титрования при использовании неводных растворителей. Они позволяют анализировать многокомпонентные системы, которые в водном растворе определить не удается, провести анализ веществ, нерастворимых или разлагающихся в воде, и т. д. Потенциометрическое титрование легко может быть автоматизировано. Промышленность выпускает несколько типов автотитраторов, использующих потенциометрические датчики. К недостаткам потенциометрического титрования можно отнести не всегда быстрое установление потенциала после добавления титранта и необходимость во многих случаях проводить при титровании большое количество отсчетов. В потенциометрическом анализе основными измерительными приборами являются потенциометры различных типов. Они предназначены для измерения ЭДС электродной системы. Так как ЭДС зависит от активности соответствующих ионов в растворе, многие потенциометры позволяют непосредственно измерять также величину рХ – отрицательный логарифм активности иона Х. Такие потенциометры в комплекте с соответствующим ионоселективным электродом носят название иономеров. Если потенциометр и электродная система предназначены для измерения активности только водородных ионов, прибор называется рН-метром.
*39)Потенциометрическое титрование и прямая потенциометрия. Потенциометрическое титрование основано на определении точки эквивалентности по результатам потенциометрических измерений. Вблизи точки эквивалетности происходит резкое изменение (скачок) потенциала индикаторного электрода. Так же, как и в других титриметрических методах, реакции потенциометрического титрования должны протекать строго стехиометрически, иметь высокую скорость и идти до конца. Для потенциометрического титрования собирают цепь из индикаторного электрода в анализируемом растворе и электрода сравнения. В качестве электродов сравнения чаще всего используют каломельный или хлорсеребряный электроды. Тип применяемого индикаторного электрода при потенциометрическом титровании зависит от свойств титриметрической смеси и ее взаимодействия с электродом. В кислотно-основном титровании используют стеклянный электрод, в окислительно-восстановительном - инертный (платиновый) электрод или электрод, обратимый по отношению к одному из ионов, содержащихся в титриметриметрической смеси; в осадительном – серебряный электрод; в комплексонометрическом - металлический электрод, обратимый к титруемому иону металла. Для нахождения точки эквивалентности часто строят дифференциальную кривую в координатах DЕ/DV – V. На точку эквивалентности указывает максимум полученной кривой, а отсчет по оси абсцисс, соответствующий этому максимуму, дает объем титранта, израсходованного на титрование до точки эквивалентности. Определение точки эквивалентности до дифференциальной кривой значительно точнее, чем по простой зависимости Е – V. Основными достоинствами метода потенциометрического титрования являются высокая точность и возможность проводить определения в разбавленных растворах, в мутных и окрашенных средах, а также определять несколько веществ в одном растворе без предварительного разделения. Значительно расширяется область практического применения потенциометрического титрования при использовании неводных растворителей. Они позволяют анализировать многокомпонентные системы, которые в водном растворе определить не удается, провести анализ веществ, нерастворимых или разлагающихся в воде, и т. д. Потенциометрическое титрование легко может быть автоматизировано. Промышленность выпускает несколько типов автотитраторов, использующих потенциометрические датчики. К недостаткам потенциометрического титрования можно отнести не всегда быстрое установление потенциала после добавления титранта и необходимость во многих случаях проводить при титровании большое количество отсчетов. В потенциометрическом анализе основными измерительными приборами являются потенциометры различных типов. Они предназначены для измерения ЭДС электродной системы. Так как ЭДС зависит от активности соответствующих ионов в растворе, многие потенциометры позволяют непосредственно измерять также величину рХ – отрицательный логарифм активности иона Х. Такие потенциометры в комплекте с соответствующим ионоселективным электродом носят название иономеров. Если потенциометр и электродная система предназначены для измерения активности только водородных ионов, прибор называется рН-метром. *40)Хроматографические методы анализа. Классификация. Характеристики хроматограмм. Хроматография -физико-химический метод разделения и анализа смесей, основанный на распределении их компонентов между двумя фазами - неподвижной и подвижной (элюент), протекающей через неподвижную. Хроматографический анализ является критерием однородности вещества: если каким-либо хроматографическим способом анализируемое вещество не разделилось, то его считают однородным (без примесей). Плоскостная хроматография.Плоскостная хроматография подразделяется на тонкослойную и бумажную. В тонкослойной хроматографии тонкий слой гранулированного сорбента или пористая плёнка наносится на стеклянную или металлическую пластинки; в случае бумажной хроматографии используют специальную хроматографическую бумагу. Тонкослойная (ТСХ) и бумажная хроматография используются для анализа жиров, углеводов, белков и др. природных веществ и неорганических соединений. Rf – относительная скорость перемещения компонентов вдоль НФ. Величина Rf определяется экспериментально, как отношение расстояния, пройденного веществом к расстоянию, пройденному растворителем от линии старта до линии финиша. Поглощение растворенного вещества поверхностью твердых тел или жидкостью, называют сорбцией, а вещества поглотители – сорбентом. При прохождении вещества через хроматографическую колонку, сила их взаимодействия с сорбентом и растворителем различна. Одни лучше взаимодействуют с сорбентом, а другие лучше взаимодействуют с растворителем, поэтому одни будут удерживаться сорбентом, а другие лучше вымываться растворителем. Хроматография впервые была введена в аналитическую практику русским ботаником М.С. Цветом. В первых же работах с помощью этого метода М.С. Цвет установил, что считавшийся однородным зеленый пигмент растений хлорофилл на самом деле состоит из нескольких веществ. При пропускании экстракта зеленого листа через колонку, заполненную порошком мела, и промываниипетролейным эфиром он получил несколько окрашенных зон, что несомненно говорило о наличии в экстракте нескольких веществ. Впоследствии это было подтверждено другими исследователями. Этот метод он назвал хроматографией, хотя сам же указал на возможность разделения и бесцветных веществ. Вещество подвижной фазы непрерывно вступает в контакт с новыми участками адсорбента и частью адсорбируется, а адсорбированное вещество контактирует со свежими порциями подвижной фазы и частично десорбируется. Таким образом, создателю хроматографического метода был известен один механизм взаимодействия разделяемых веществ с материалом колонки – молекулярная адсорбция. М.С. Цвет сформулировал закон, который назвал законом адсорбционного замещения: Вещества, растворенные в определенной жидкости, образуют определенный адсорбционный ряд А, В, С,…, выражающий относительное адсорбционное сродство его членов к адсорбенту. Каждый из членов адсорбционного ряда, обладая большим адсорбционным сродством, чем последующий, вытесняет его из соединения и в свою очередь вытесняется предыдущим. Таким образом, основным условием для осуществления хроматографического процесса – процесса разделения веществ на колонке – М.С. Цвет считал различие в адсорбируемости. В современной хроматографии для разделения веществ кроме молекулярной адсорбции используют и другие физико-химические явления. Имеется несколько классификаций, основанных на различных принципах. Общепринятыми являются следующие. По агрегатному состоянию применяемых фаз. Согласно этой классификации хроматографию подразделяют на газовую и жидкостную. Газовая включает газо-жидкостную и газо-адсорбционную хроматографию. Жидкостная хроматография подразделяется на жидкостно – жидкостную, жидкостно – адсорбционную и жидкостно – гелевую. Первое слово в этой классификации характеризует агрегатное состояние подвижной фазы. По механизмам разделения, т.е. по характеру взаимодействия между сорбентом и сорбатом. По этой классификации хроматографию подразделяют на следующие виды: 1. адсорбционная хроматография – разделение основано на различии в адсорбируемости разделяемых веществ твердым адсорбентом; 2. распределительная хроматография – разделение основано на различии в растворимости разделяемых веществ в неподвижной фазе (газовая хроматография) и на различии в растворимости разделяемых веществ в подвижной и неподвижной жидких фазах; 3. ионообменная хроматография – разделение основано на различии в способности разделяемых веществ к ионному обмену; 4. проникающая хроматография – разделение основано на различии в размерах или формах молекул разделяемых веществ, например, при применении молекулярных сит (цеолитов); 5. осадочная хроматография – разделение основано на образовании различных по растворимости осадков разделяемых веществ с сорбентом; 6. адсорбционно-комплексообразовательная хроматография – разделение основано на образовании координационных соединений различной прочности в фазе или на поверхности адсорбента. Следует иметь в виду, что очень часто процесс разделения протекает по нескольким механизмам. По применяемой технике: 1) колоночная хроматография – разделение веществ проводится в специальных колонках; 2) плоскостная хроматография: а – бумажная – разделение веществ проводится на специальной бумаге; б – тонкослойная – разделение веществ проводится в тонком слое сорбента. В колоночной и тонкослойной хроматографии можно использовать любой из приведенных выше механизмов разделения, в бумажной хроматографии чаще всего применяют распределительный и ионообменный механизмы. По способу относительного перемещения фаз различают фронтальную, или элюэнтную, и вытеснительную хроматографию. *41)Обзор методов окислительно-восстановительного титрования. Определение конечной точки. Погрешности. Перманганатометрия - метод, который основан на окислительной способности рабочего раствора перманганата калия KМnO4. Титрование ведется без индикатора. Применяется для определения только восстановителей при прямом титровании.Иодометрия – метод, в котором рабочим титрованным раствором служит раствор свободного иода в КI. Метод позволяет определять как окислители, так и восстановители. Индикатором служит крахмал.Дихроматометрия основана на использовании в качестве рабочего раствора дихромата калия K2Cr2O7. Метод может применяться как для прямых так и косвенных определений восстановителей.Броматометрия основана на использовании в качестве титранта бромата калия KBrO3 при определении восстановителей.Иодатометрия применяет в качестве рабочего раствора раствор иодата калия KIO3 при определении восстановителей.Ванадатометрия дает возможность использовать окислительную способность ванадата аммоноя NH4VO3. Кроме перечисленных методов в лабораторной практике используются и такие методы как цериметрия (Ce4+), титанометрия и другие.Для вычисления молярной массы эквивалента окислителей или восстановителей учитывается число электронов, принимающих участие в окислительно-восстановительной реакции (Мэ = М/ne , где n – число электронов е). Для определения числа электронов необходимо знать начальную и конечную степень окисления окислителя и восстановителя.
Из большого числа окислительно-восстановительных реакций для химического анализа используют только те реакции, которые:
· протекают до конца;
· проходят быстро и стехиометрично;
· образуют продукты определенного химического состава (формулы);
· позволяют точно фиксировать точку эквивалентности;
· не вступают в реакцию с побочными продуктами, присутствующими в исследуемом растворе.
Наиболее важными факторами, оказывающими влияние на скорость реакции, являются:
· концентрация реагирующих веществ;
· температура;
· значение рН раствора;
· присутствие катализатора.
В большинстве случаев скорость реакции находится в прямой зависимости от температуры и рН раствора. Поэтому многие определения методом окислительно-восстановительного титрования следует проводить при определенном значении рН и при нагревании.Индикаторы окислительно-восстановительного титрования.окислительный восстановительный титрование.При анализе методами окислительно-восстановительного титрования используется прямое, обратное и заместительное титрование. Точка эквивалентности окислительно-восстановительного титрования фиксируется как с помощью индикаторов, так и безиндикаторным способом. Безиндикаторный способ применяется в тех случаях, когда окисленная и восстановленная формы титранта отличаются. В точке эквивалентности, при введении 1 капли избытка раствора титранта изменит окраску раствора. Безиндикаторным способом можно проводить определения перманганатометрическим методом, т.к. в точке эквивалентности от одной капли раствора перманганата калия титруемый раствор окращивается в бледнорозовый цвет.При индикаторном способе фиксирования точки эквивалентности применяют специфические и редоксиндикаторы. К специфическим индикаторам относится крахмал в иодометрии, который в присутствии свободного иода окрашивается в интенсивно-синий цвет вследствие образования адсорбционного соединения синего цвета. Редокс-индикаторы – это вещества, у которых окраска меняется при достижении определенного значения окислительно-восстановительного (редокспотенциала). К редокс-индикаторам относится, например, дифениламин NH(C6H5)2. При действии на бесцветные растворы его окислителями он окрашивается в сине-фиолетовый цвет.
Редокс-индикаторам предъявляют следующие требования:
· окраска окисленной и восстановленной формы должна быть различна;
· изменение цвета должно быть заметно при небольшом количестве индикатора;
· индикатор должен реагировать в точке эквивалентности с весьма небольшим избытком восстановителя или окислителя;
· интервал действия его должен быть как можно меньше;
· индикатор должен быть устойчив к воздействию компонентов окружающей среды (О2, воздуха, СО2, света и т.п.).
Интервал действия редокс-индикатора рассчитывается по формуле:
Е = Ео ± 0,058/n ,
где Ео - нормальный окислительно-восстановительный потенциал индикатора (в справочнике), n - число электронов, принимающих в процессе окисленияили восстановления индикатора.
Перманганатометрия.В основе перманганатометрии лежит реакция окисления различных восстановителей рабочим раствором перманганата калия, т.е. ионом MnO4-. Окисление перманганатом калия можно проводить в кислой, нейтральной и в щелочной среде
В сильнокислой среде перманганат-ионы (МnО4-) обладают высоким окислительно-восстановительным потенциалом, восстанавливаясь до Мn2+, и их применяют для определения многих восстановителей:
МnО4- + 8Н+ + 5е = Мn2+ + 4Н2О
Е0 МnО4- / Мn2+ = 1,51 В
В щелочной среде МnО4- восстанавливается до манганат иона:
МnО4- + е = МnО42-
В нейтральной или слабощелочной среде перманганат ион восстанавливается до марганцовистой кислоты MnO(OH)2 или до MnO2:
МnО4- + 2Н2О + 3е = МnО2↓ + 4ОН-
Е0 МnО4- / МnО2 = 0,59 В
При титровании перманганатом не применяют индикаторы, так как реагент сам окрашен и является чувствительным индикатором: 0,1 мл 0,01М раствора КМnО4 окрашивает 100 мл воды в бледно-розовый цвет. В результате реакции перманганата калия с восстановителем в кислой среде образуются бесцветные ионы Мn2+, что позволяет четко фиксировать точку эквивалентности.Раствор КМnО4 относится к титрантам с установленным титром. В связи с этим перед использованием его в анализе в качестве титранта раствор КМnО4 стандартизируют по концентрации растворов исходных веществ шавелевой кислоты или оксалата натрия. Раствор перманганата калия очень трудно получить в чистом виде. Обычно он загрязнен следами оксида марганца (IV). Кроме того, чистая дистиллированная вода обычно содержит следы веществ, которые восстанавливают перманганат калия с образованием оксида марганца (IV):
4 КМnО4 + 2Н2О = 4 МnО2↓ + 4ОН- + 3О2
При хранении в твердом виде перманганат калия разлагается под действием света, загрязняясь также МnО2:
КМnО4 = К2МnО4 + МnО2↓ + О2
Раствор перманганата калия может быть приготовлен из стандарт - титра и по навеске взятой на технических весах. В первом случае содержимое ампулы количественно переносится в мерную колбу вместимостью 2л, опаласкивая ампулу и воронку теплой дистиллированной водой. Внести в мерную колбу небольшой объем горячей воды для растворения кристаллов, затем полученный раствор охладить до комнатной температуры, объем раствора довести до метки и пермешать. Молярная концентрация полученного раствора составляет 0,05 моль/л.Во втором случае на технических весах в бюксе или на часовом стекле отвесить навеску перманганата калия массой 1,6 г, поместить ее в химический стакан и растворять в горячей дистиллированной воде при тщательном перемешивании образующегося раствора, стараясь, чтобы все кристаллы К
|
из
5.00
|
Обсуждение в статье: Классификация методов комплексонометрического титрования |
|
Обсуждений еще не было, будьте первым... ↓↓↓ |

Почему 1285321 студент выбрали МегаОбучалку...
Система поиска информации
Мобильная версия сайта
Удобная навигация
Нет шокирующей рекламы