 |
Главная |


Строение и классификация галогенопроизводных
|
из
5.00
|
Атомы галогенов связаны с атомом углерода одинарной связью. Как и другие органические соединения, строение галогенопроизводных может быть выражено несколькими структурными формулами, например:
Классифицировать галогенопроизводные можно несколькими способами:
1) в соответствии с общей классификацией углеводородов (т.е. алифатические, алициклические, ароматические, предельные или непредельные галогенопроизводные)
2) по количеству и качеству атомов галогенов
3) по типу атома углерода, к которому присоединён атом галогена: первичные, вторичные, третичные галогенопроизводные.
Номенклатура галогенуглеводородов
Согласно правилам ИЮПАК[1] галогенуглеводороды рассматриваются как продукты замещения углеводородов, соответствующие атомы галогенов указываются в префиксе. Если это необходимо положение атома галогена указывается цифрой.
Физические свойства
За исключением низших гомологов (кроме иодалканов), газообразных при нормальных условиях, галогенуглеводороды представляют собой бесцветные жидкости итвёрдые вещества со своеобразным запахом (сладковатым для алифатических соединений). Полииодалканы окрашены в жёлтый цвет. В гомологическом ряду наблюдаются такие же изменения, как и у незамещённых углеводородов. Так, с удлинением цепи и ростом молекулярной массы растёт и температура кипения, а при увеличении степени разветвлённости температуры кипения падают. Однако полифторалканы с бо́льшим количеством атомов фтора в молекуле кипят ниже прочих — причиной этому, видимо, является уменьшение межмолекулярного взаимодействия.
Хотя алкилгалогениды и полярные соединения (дипольные моменты моногалогеналканов примерно равны величине μ у воды – ср. μ(H2O) = 1,85 D, а μ(CH3Cl) = 1,9 D), но в воде они нерастворимы, вероятно, из-за того, что они не способны образовывать полярные связи. Они растворимы в обычных органических растворителях.
Стоит отметить, что полярность связи углерод—галоген зависит не только от разницы электроотрицательностей атомов, но и от наличия неподелённых пар электронову атома галогена, а также от сравнительных ковалентных радиусов атомов. Это весьма очевидно, если рассматривают связи C—F и C-Cl: их дипольные моменты равны, соответственно, 1,81 и 1,83 D (лучшее сопряжение для связи C—F)!
Ввиду полярности связи C–X атом улерода является несколько электрофильным (см., например, статью "Реакции электрофильного замещения"). Неподелённыеэлектронные пары атома галогена придают молекуле также слабый электронодонорный характер (особенно иодуглеводородам). Полярность связи C–X падает при переходе от атома углерода в sp3–гибридизации к атому углерода в sp—гибридизации. Например, дипольные моменты хлорэтана, хлорэтена, хлорбензола и хлорэтинасоставляют соответственно 2,0, 1,44, 1,58 и 0,44 D. Причина тому — возрастает электроотрицательность атома углерода, а неподелённая электронная пара вступает в сопряжение с π-электронной системой алкена, арена (положительный мезомерный (+M) зффект). Поскольку одновременно имеет место отрицательный индуктивный (-I) эффект, трудно предугадать распределение электронной плотности в системе. Расчёты для простейших соединений (аллилгалогенид, фенилгалогеноид) показывают, что непосредственно связанный с атомом галогена углеродный атом имеет некоторый положительный заряд, а сам атом галогена и атом углерода по другую сторону двойной связи (в случае аренов — о- и (в меньшей степени) п-места) заряжены несколько отрицательно. Возрастают немного и электронодонорные свойства π-системы (ср. энергия ионизации этилена и винилхлорида: 10,5 и 10 эВ соответственно).
Получение
При синтезе галогеналканов обычно исходят из углеводородов, спиртов или карбоновых кислот.
Получение галогеналканов
Прямое галогенирование
Радикальным галогенированием можно получать хлор- или бромалканы. Недостатком этого метода является то, что образуется смесь различных продуктов замещения. При этом наряду с изомерными монозамещёнными в смеси также содержатся ди- и полизамещённые соединения. К тому же, например, эквимолярная смесь Cl2 и CH4взрывоопасна, не говоря уже о смесях алканов со фтором. Однако меняя условия процесса можно добиться приемлемых для промышленности выходов. Например, при хлорировании алкан берут в избытке. Продукты разделяют фракционной перегонкой. К примеру, так в промышленности получают метиленхлорид итетрахлоруглерод.
Алканы хлорируются при интенсивном УФ-облучении или нагревании (обычно применяют первое). Наиболее легко образуются третичные радикалы и, соответственно, галогеналканы; наименее – первичные. Бромирование мало характерно для алканов легче гексана, а прочие бромируются при одновременном освещении и кипячении.
CnH2n+2 + X2  CnH2n+1X + HX
CnH2n+1X + HX 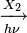 CnH2nX2 + HX ...
CnH2nX2 + HX ...
В присутствии специальных реагентов (например, N-Бромсукцинимида, сокращённо NBS), а также инициаторов свободных радикалов (таких, как УФ-облучение, нагревание, пероксиды) замещённые алкены бромируются в аллильное положение.

Аллильное хлорирование осуществляется лишь при 400–600°C, обычно же идут конкурирующие реакции – присоединение по двойной связи (в этих условиях — тоже по радикальному механизму), полимеризация, изомеризация алкенов. В промышленности так производят аллилхлорид.
Замещённые арены хлорируются и бромируются в боковую цепь. Например, толуол при нагревании и интенсивном освещении хлорируют, получая бензилхлорид.
Радикальным методом получают также и перфторалканы. Реакция протекает очень энергично, и вследствие большого тепловыделения приходится разбавлять фторазотом, применяют также медные сетки для отвода теплоты. В процессе применяют переносчики фтора – фториды металлов, как CoF2, MnF2, AgF, образующие в ходе реакции (при нагревании) соответственно CoF3, MnF4, AgF2.
Получение моногалогеналканов
1. Присоединение галогеноводородов к алкенам.
R-CH=CH2+HCl→R-CHCl-CH3
См. также: Реакции электрофильного присоединения
Весьма легко присоединяется фтор (трудноуправляемый процесс, возможен взрыв), иод присоединяется медленно. Обычно происходит стереоселективно, кроме как в присутствии свободных радикалов. В присутствии других нуклеофилов возможно сопряжённое присоединение (см. статью Галогенгидрины).
1. Реакции спиртов с галогеноводородами.
R-OH+H-Cl→R-Cl+H2O
2. Взаимодействие галогенидов фосфора или тионилхлорида со спиртами.
3R-OH+PCl3→3R-Cl+H3PO3
3. Реакция Бородина — Хунсдикера
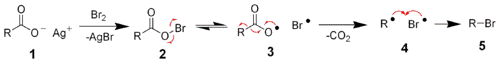
1. Реакция Сварта
R-Cl+AgF→R-F+AgCl
Получение дигалогеналканов
1. Присоединение галогеноводородов к алкинам.
R-C≡CH+2HCl→R-CCl2-CH3
2. Взаимодействие альдегидов и кетонов с PCl5, PBr5 или SF4. Реакция идёт при нагревании.
R—CHO + PCl5 → R—CHCl2,
3. Присоединение галогенов к алкенам
R-CH=CH2 + Cl2 → R-CHCl-CHCl
4. Раскрытие циклических простых эфиров (например, ТГФ) при реакции с NaI в среде H3PO4+P2O5.
C4H8O + HI → I-CH2CH2CH2CH2-I
При 180 °C ТГФ с хлороводородом даёт 1,4-дихлорбутан.
Получение галогеналкенов
1. С алкинами напрямую — как H-электрофилы галогеноводороды взаимодействуют медленно. Реакцию значительно ускоряют соли меди (I) и ртути (II); по сути, происходит нуклеофильное присоединение галогенид-иона. Например, есть такой способ производства винилхлорида в газовой фазе на активированном угле исулеме при 120—180 °C.
2. Также и галогены присоединяются по тройной связи медленнее, чем по двойной. Вероятно, реакция идёт через КПЗ, который опять-таки является уже электрофильной частицей.
Или применяют производные алкенов. В частности, для получения того же винилхлорида есть такие два способа.
1. Дегидратация этиленхлоргидрина на катализаторе.
1. Дегидрохлорирование дихлорэтана в газовой фазе при 400—500 °C
Получение галогенаренов
1. Галогенирование аренов или алкиларенов в ядро
Ph-H+Cl2→Ph-Cl
2. Разложение арендиазониевых солей
Ph-N+≡N Cl−→Ph-Cl
Получение бензилгалогенидов
1. Галогенирование алкиларенов в боковую цепь.
Ph-CH3+Cl2→Ph-CH2Cl
2. Хлорметилирование (р-ция Блана)
Ph-H+HCHO+HCl→Ph-CH2Cl
Химические свойства
Реакционная способность галогенуглеводородов главным образом зависит от поляризуемости связи C-X, падающей в ряду C-I > C-Br > C-Cl >> C-F
Также весьма существенный вклад в реакционную способность вносит строение алкильного радикала, наличие кратных связей, разветвлённость углеводородного скелета, возможность делокализации заряда у α-атома.
Реакции галогенуглеводородов
Основные реакции галогенуглеводородов
1)Нуклеофильное замещение
1. R-X+NH3→R-NH2+R2NH+R3N+R4N+X−
2. R-X+NO2−→R-NO2
3. R-X+CN−→R-CN
4. R-X+OH−→R-OH
5. R-X+R1-O−→R-O-R1
6. R-X+R1-COO−→R-OCOR1
7. R-X+SCN−→R-SCN
2)Дегидрогалогенирование (элиминирование)
R-CH2-CHCl+OH−→R-CH=CH2
3)Синтез реактива Гриньяра
R-X+Mg→RMgX
4)Восстановление
R-X+H2→R-H+HX
Галогенопроизводные углеводородов являются наиболее важными промежуточными соединениями в разнообразных органических синтезах, а значительная доля превращений, происходящих с галогенопроизводными, состоит в нуклеофильном замещении галогена. Для этого у галогенопроизводных имеются определенные предпосылки сильнаяполяризация связи С—Hal и большая подвижность галогена как уходящей группы. Однако в зависимости от строения углеводородного радикала R в молекуле R—Hal скорость реакции нуклеофильного замещения может варьировать в широких пределах.[c.170]
В настоящее время органические перекиси применяются в промышленности в качестве полупродуктов и как инициаторы полимеризации. Несомненно, что началом такого широкого распространения перекисей в химической промышленности послужилопроизводство фенола и ацетона через кумилгидроперекись. Представляется возможным осуществить аналогичным путем производство замещенных фенолов и двухатомных фенолов— гидрохинона, резорцина и пирокатехина однако данных о получении этихсоединений в крупном масштабе еще нет. Кроме того, перекиси используются также в качестве катализаторов некоторых процессовсинтеза, в особенности таких, которые могут рассматриваться какограниченная полимеризация, например в реакциях галогенопроизводных углеводородов или альдегидов с олефинами, и в других реакциях тело-меризации.
Спирты
Спирты́ (от лат. spiritus — дух; устар. алкого́ли, от араб. الكحول аль-кухуль — порошок[1]) — органические соединения, содержащие одну или более гидроксильных групп (гидроксил, −OH), непосредственно связанных с насыщенным (находящимся в состоянии sp³-гибридизации) атомом углерода[2]. Спирты можно рассматривать как производные воды (H−O−H), в которых один атом водородазамещен на органическую функциональную группу: R−O−H.
В номенклатуре ИЮПАК для соединений, в которых гидроксильная группа связана с ненасыщенным (sp²-гибридным) атомом углерода, рекомендуются названия «енолы» (гидроксил связан с винильной C=C-связью)[3] и «фенолы» (гидроксил связан сбензольным или другим ароматическим циклом)[4].
Спирты представляют собой обширный и разнообразный класс соединений: они весьма распространены в природе  и часто выполняют важные функции в живых организмах . Спирты являются важными соединениями с точки зрения органического синтеза, не только представляя интерес как целевые продукты, но и как промежуточные вещества, имеющие ряд уникальных химических свойств . Кроме того, спирты являются промышленно важными продуктами и находят широчайшее применение как в промышленности, так и в повседневных приложениях .
и часто выполняют важные функции в живых организмах . Спирты являются важными соединениями с точки зрения органического синтеза, не только представляя интерес как целевые продукты, но и как промежуточные вещества, имеющие ряд уникальных химических свойств . Кроме того, спирты являются промышленно важными продуктами и находят широчайшее применение как в промышленности, так и в повседневных приложениях .
Классификация спиртов
Спирты классифицируются следующим образом (в скобках приведены примеры)[8]:
1. По числу гидроксильных групп:
— одноатомные спирты (метанол);
— двухатомные спирты (этиленгликоль);
— трёхатомные спирты (глицерин);
— четырёхатомные спирты (пентаэритрит);
— многоатомные спирты (пятиатомный спирт: ксилит).
1. В зависимости от насыщенности углеводородного заместителя:
— предельные (насыщенные) спирты (бутанол);
— непредельные (ненасыщенные) спирты (аллиловый спирт, пропаргиловый спирт);
— ароматические спирты (бензиловый спирт).
1. В зависимости от наличия или отсутствия цикла в углеводородном заместителе:
— ациклические (алифатические) спирты (этанол);
— алициклические спирты (циклогексанол).
1. В зависимости от числа заместителей при α-углеродном атоме:
— первичные спирты (этанол);
— вторичные спирты (пропанол-2);
— третичные спирты (2-метилпропанол-2).
Номенклатура спиртов
Систематическая номенклатура
По номенклатуре ИЮПАК названия простых спиртов образуются от названий соответствующих алканов с добавлением суффикса «-ол», положение которого указывается арабской цифрой.
Правила построения названий спиртов[9]:
1. Выбирают родительский углеводород по самой длинной непрерывной углеводородной цепи, содержащей гидроксильную группу. Он формирует базовое название (по числу атомов углерода).
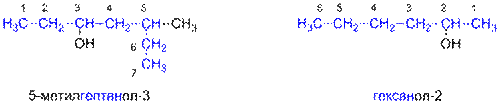
2. Родительский углеводород нумеруют в таком направлении, чтобы гидроксильная группа получила наименьший номер в названии. (Если в соединении имеются функциональные группы старше гидроксильной, то это правило применяется к старшей функциональной группе.)
3. Старшая функциональная группа обозначается в виде суффикса (для гидроксильной — -ол), а остальные заместители — в виде приставок в алфавитном порядке. Их положение в углеводородной цепи обозначается при помощи цифр — локантов, помещаемых после суффиксов и перед приставками[10]. Для многоатомных спиртов перед суффиксом -ол указывается число гидроксильных групп (-диол, -триол, -тетраол и т. д.).

4. Если при различных вариантах нумерации цепи гидроксильная группа получает один и тот же локант, то цепь нумеруют в том направлении, при котором другой заместитель получает наименьший локант.
Получение одноатомного спирта
1. Самый общий способ получения спиртов, имеющий промышленное значение, — гидратация алкенов. Реакция идет при пропускании алкена с парами воды над фосфорнокислым катализатором:
H3PO4
СН2=СН2 + Н2О → СН3—СН2—ОН.
Из этилена получается этиловый спирт, из пропена — изопропиловый. Присоединение воды идет по правилу Марковникова, поэтому из первичных спиртов по данной реакции можно получить только этиловый спирт.
2. Другой общий способ получения спиртов — гидролиз алкилгалогенидов под действием водных растворов щелочей:
R—Br + NaOH → R—OH + NaBr.
По этой реакции можно получать первичные, вторичные и третичные спирты.
3. Восстановление карбонильных соединений. При восстановлении альдегидов образуются первичный спирты, при восстановлении кетонов — вторичные:
R—CH=O + Н2 → R—CH2—OH, (1)
R—CO—R' + Н2 → R—CH(OH) —R'. (2)
Реакцию проводят, пропуская смесь паров альдегида или кетона и водорода над никелевым катализатором.
4. Действие реактивов Гриньяра на карбонильные соединения .
5. Этанол получают при спиртовом брожении глюкозы:
С6Н12О6 → 2С2Н5ОН + 2СО2↑.
Химические свойства спиртов определяются присутствием в их молекулах гидроксильной группы ОН. Связи С-О и О-Н сильно полярны и способны к разрыву. Различают два основных типа реакций спиртов с участием функциональной группы -ОН:
1) Реакции с разрывом связи О-Н: а) взаимодействие спиртов с щелочными и щелочноземельными металлами с образованием алкоголятов; б) реакции спиртов с органическими и минеральными кислотами с образованием сложных эфиров; в) окисление спиртов под действием дихромата или перманганата калия до карбонильных соединений. Скорость реакций, при которых разрывается связь О-Н, уменьшается в ряду: первичные спирты > вторичные > третичные.
2) Реакции сопровождающиеся разрывом связи С-О: а) каталитическая дегидратация с образованием алкенов (внутримолекулярная дегидратация) или простых эфиров (межмолекулярная дегидратация): б) замещение группы -ОН галогеном, например при действии галогеноводородов с образованием алкилгалогенидов. Скорость реакций, при которых разрывается связь С-О, уменьшается в ряду: третичные спирты > вторичные > первичные.
Спирты являются амфотерными соединениями.
Реакции с разрывом связи О-Н.
1. Кислотные свойства спиртов выражены очень слабо. Низшие спирты бурно реагируют со щелочными металлами:
2С2Н5-ОН + 2K→ 2С2Н5-ОK + Н2↑, (3)
но не реагируют с щелочами. С увеличением длины углеводородного радикала скорость этой реакции замедляется.
В присутствии следов влаги соли спиртов (алкоголяты) разлагаются до исходных спиртов:
С2Н5ОK + Н2О → С2Н5ОН + KОН.
Это доказывает, что спирты — более слабые кислоты, чем вода.
2. При действии на спирты минеральных и органических кислот образуются сложные эфиры. Образование сложных эфиров протекает по механизму нуклеофильного присоединения-отщепления :
Н+
С2Н5ОН + СН3СООН  СН3СООС2Н5 + Н2О
СН3СООС2Н5 + Н2О
Этилацетат
C2H5OH + HONO2 C2H5ONO2 + Н2O
Этилнитрат
Отличительной особенностью первой из этих реакций является то, что атом водорода отщепляется от спирта, а группа ОН - от кислоты. (Установлено экспериментально методом "меченых атомов" ).
3. Спирты окисляются под действием дихромата или перманганата калия до карбонильных соединений. Первичные спирты окисляются в альдегиды, которые, в свою очередь, могут окисляться в карбоновые кислоты:
[O] [О]
R-CH2-OH → R-CH=O → R-COOH.
Вторичные спирты окисляются в кетоны:

Третичные спирты могут окисляться только с разрывом С-С связей.
Реакции с разрывом связи С-О.
1) Реакции дегидратации протекают при нагревании спиртов с водоотнимающими веществами. При сильном нагревании происходит внутримолекулярная дегидратация с образованием алкенов:
H2SO4 ,t >150°С
СН3-СН2-СН2-ОН → СН3-СН=СН2 + Н2О.
При более слабом нагревании происходит межмолекулярная дегидратация с образованием простых эфиров:
H2SO4,t< 150°С
2CH3-CH2-OH → C2H5-O-C2H5 + H2O.
2) Спирты обратимо реагируют с галогеноводородными кислотами (здесь проявляются слабые основные свойства спиртов):
ROH + HCl RCl + Н2О
Третичные спирты реагируют быстро, вторичные и первичные - медленно.
Физические свойства и строение спиртов
Молекулы спиртов, подобно молекуле воды, имеют угловое строение. Угол R−O−H в молекуле метанола равен 108,5°[55]. Атом кислородагидроксильной группы находится в состоянии sp³-гибридизации. Спирты имеют существенно более высокие температуры плавления и кипения, чем можно было бы предполагать на основании физических свойств родственных соединений. Так, из ряда монозамещённых производных метана, метанол имеет необычно высокую температуру кипения, несмотря на относительно небольшую молекулярную массу[56]:
Молекулярные массы и температуры кипения метана и некоторых его производных[57]
| Метан CH4 | Метанол CH3OH | Хлорметан CH3Cl | Нитрометан CH3NO2 | Бромметан CH3Br | |
| Молярная масса, г/моль | 16,04 | 32,04 | 50,48 | 61,04 | 94,94 |
| Температура кипения, °С | −161,5 | 64,5 | −24,2 | 101,2 | 3,6 |
Высокие температуры кипения спиртов объясняются наличием межмолекулярных водородных связей[55]. Энергия водородной связи значительно ниже, чем энергия ковалентной химической связи. Так, например, для метанола энергия водородной связи составляет 16,7 кДж/моль[58] , тогда как связи C–H, C–O и O–H имеют энергию 391,7, 383,5 и 428,8 кДж/моль соответственно[59]. Тем не менее, влияние водородных связей на физические свойства спиртов весьма значительное.
Молекулы спирта, имея две полярных связи C−O и O−H, обладают дипольным моментом (~5,3—6,0·10−30 Кл·м)[55]. Электростатические заряды в молекуле метанола составляют: на атоме углерода 0,297 e; на атоме гидроксильного водорода 0,431 e; на атоме кислорода −0,728 e[60]. Вместе с тем, энергия ионизации спиртов ниже, чем у воды (10,88 эВ для метанола против 12,61 эВ для воды)[61], что объясняется электронодонорным эффектом алкильной группы.
Следует отметить, что влияние гидроксильной группы особенно велико на соединения с небольшой углеводородной цепочкой. Так, например, метанол и этанол неограниченно смешиваются с водой и имеют довольно высокие плотности и температуры кипения для своей молекулярной массы, в то время как высшие спирты гидрофобны и мало отличаются по свойствам от соответствующих углеводородов[62].
Фенолы.
Фено́лы — органические соединения ароматического ряда, в молекулах которых гидроксильные группы OH− связаны с атомамиуглерода ароматического кольца.
Классификация
По числу ароматических ядер различают собственно фенолы, нафтолы (2 конденсированных ядра), антролы (3 ядра), фенантролы (4 ядра), бензотетролы (5 ядер),
По числу ОН-групп различают:
1. одноатомные фенолы (аренолы): фенол (C6H5OH) и его гомологи;
2. двухатомные фенолы (арендиолы): гидрохинон, пирокатехин, резорцин (соответственно 1,2-, 1,3- и 1,4-гидроксибензолы);
3. трёхатомные фенолы (арентриолы): пирогаллол, гидроксигидрохинон, флороглюцин (соответственно 1,2,3-, 1,2,4- и 1,3,5-тригидроксибензолы),
4. многоатомные фенолы
Изомерия
Возможны 2 типа изомерии:
1. изомерия положения заместителей в бензольном кольце;
2. изомерия боковой цепи (строения алкильного радикала и числа радикалов).
Электронное строение[править | править вики-текст]
Фенолы представляют собой полярные соединения (диполи). Бензольное кольцо является отрицательным концом диполя, группа — OH — положительным. Дипольный момент направлен в сторону бензольного кольца.

Электронное строение фенола
Как известно, гидроксильная группа -OH является заместителем I рода, то есть она способствует повышению электронной плотности в бензольном кольце (особенно в орто- и пара-положениях). Это обусловлено тем, что одна из неподелённых пар электронов атома кислорода OH-группы вступает в сопряжение с π-системой бензольного кольца. Смещение неподелённой пары электронов атома кислорода в сторону бензольного кольца приводит к увеличению полярности связи O-H. Таким образом, имеет место взаимное влияние атомов и атомных групп в молекуле фенола. Это взаимное влияние отражается в свойствах фенола.[1]
Во-первых, повышается способность к замещению атомов водорода в орто- и пара-положениях бензольного ядра, и в результате реакций замещения обычно образуются три-замещённые производные фенола.
Во-вторых, увеличение полярности связи O-H под действием бензольного ядра и появление достаточно большого положительного заряда на атоме водорода приводит к тому, что молекулы фенола диссоциируют в водных растворах по кислотному типу.
Фенол является слабой кислотой. В этом состоит главное отличие фенолов от спиртов, которые являются неэлектролитами.
Физические свойства
Большинство одноатомных фенолов при нормальных условиях представляют собой бесцветные кристаллические вещества с невысокой температурой плавления и характерным запахом. Фенолы малорастворимы в воде, хорошо растворяются в органических растворителях, токсичны, при хранении на воздухе постепенно темнеют в результате окисления. Фенол C6H5OH (карболовая кислота) — бесцветное кристаллическое вещество на воздухе окисляется и становится розовым, при обычной температуре ограниченно растворим в воде, выше 66 °C смешивается с водой в любых соотношениях. Фенол — токсичное вещество, вызывает ожоги кожи, является антисептиком!
В живых организмах
Фенол является окончанием боковой группы стандартной аминокислоты тирозина, и поэтому входит в состав практически каждой белковой молекулы[2].
Химические свойства
1. Реакции с участием гидроксильной группы
Кислотные свойства
1. Диссоциация в водных растворах с образованием фенолят-ионов и ионов водорода;
2. Взаимодействие со щелочами с образованием фенолятов (отличие от спиртов);
3. Взаимодействие с активными металлами с образованием фенолятов (образующиеся в результате реакций 2 и 3) феноляты легко разлагаются при действии кислот. Даже такая слабая кислота, как угольная, вытесняет фенол из фенолятов, следовательно, фенол — ещё более слабая кислота, чем угольная).
При взаимодействии фенолятов с галогенпроизводными образуются простые и сложные эфиры (реакция Фриделя — Крафтса).
2. Реакции с участием бензольного кольца
Реакции замещения
1. Галогенирование (взаимодействие с галогенами)
2. Нитрование (взаимодействие с азотной кислотой)
3. Сульфирование (взаимодействие с серной кислотой)
Реакции присоединения
1. Гидрирование (восстановление водородом до циклогексанола)
|
из
5.00
|
Обсуждение в статье: Строение и классификация галогенопроизводных |
|
Обсуждений еще не было, будьте первым... ↓↓↓ |

Почему 1285321 студент выбрали МегаОбучалку...
Система поиска информации
Мобильная версия сайта
Удобная навигация
Нет шокирующей рекламы