 |
Главная |


Направленность реакции
|
из
5.00
|
Закон действия масс позволяет определять не только равновесность раствора, но и направленность самопроизвольных химических реакций в нём. Для этого достаточно сопоставить фактическое произведение активностей (концентраций) участвующих в реакции компонентов -  с соответствующей условиям константой равновесия
с соответствующей условиям константой равновесия  .
.
Если природная вода имеет неравновесный состав, то  ¹
¹  . Разность между этими величинами характеризует степень неравновесности, которую можно выразить величиной свободной энтальпии:
. Разность между этими величинами характеризует степень неравновесности, которую можно выразить величиной свободной энтальпии:

 . (II‑107)
. (II‑107)
Величина ΔZr,j в этом уравнении характеризует ту максимальную полезную работу, которую должны совершить компоненты реакции j, чтобы достичь равновесного состояния, т.е. завершить релаксацию. Величина Δξj определяет то количество молей наименее затратного компонента, которое необходимо перевести из реагентов в продукты.
Если работу, которую надо совершить для достижения равновесия отнести к молю (Δξj = 1 молю), то получится уравнение Вант-Гоффа (Van't-Hoff equation):
 , (II‑108)
, (II‑108)
Это уравнение часто называют также просто уравнением изотермыреакции (isotherm equation),так каконо определяет степень неравновесности системы и направленность реакции при постоянной температуре. Для оценки направленности реакций пользуются разными величинами: lg(Пa/Kr,j) - индекс насыщения (saturation index, SIj), lg(Kr,j/Пa) – индекс неравновесности (disequilibrium index)или Пa/Kr,j = Ωj - степень насыщения (saturation state). При степени насыщения более 1 раствор считается перенасыщенным, а менее 1 – недонасыщенным относительно продуктов реакции.
Из этого уравнения II-102 следует, что
 (II‑109)
(II‑109)
В 1922 Т. де Донде (1870-1957) предложил использовать производную свободной энтальпии по глубине реакции:
 , (II‑110)
, (II‑110)
в качестве характеристики химического сродства (chemical affinity). Его величина характеризует изменение максимальной полезной работы с каждым шагом реакции в 1 моль и измеряется в Дж/моль. Положительные значения химического сродства Ar свидетельствуют об избытке реагентов и о направленности реакции слева направо. Отрицательные значения Ar,j, наоборот, свидетельствуют об избытке продуктов и направленности реакции справа налево. При равновесии в реакции Ar,j = 0.
Если всех компонентов в растворе достаточно для достижения равновесия реакции j, то конечные содержания этих компонентов в растворе стабилизируются на величине Neq,ji. Тогда максимальное значение Δξj в уравнении II-102 будет равно:
 . (II‑111)
. (II‑111)
где N0,ij– содержание компонента i реакции j до ее начала. Это уравнение позволяет определять относительную величину степени завершенности реакции (complement reaction degree):
 . (II‑112)
. (II‑112)
где Nt,ij – содержания компонента i реакции j, в некоторый момент времени t на пути релаксации.
Если для достижения равновесия не хватает какого-нибудь компонента i, то он считается лимитирующим, так как равновесие в этом случае недостижимо. Тогда степень завершенности реакции определяется по его содержанию. Если к началу реакции содержание лимитирующего компонента равно Nlim,ij, то
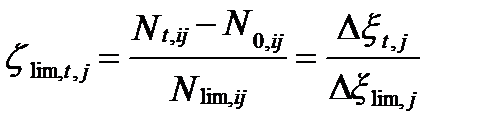 . (II‑113)
. (II‑113)
Величины степени завершенности реакции ξt,j безразмерные, меняются от 0 до 1 и позволяют уйти от экстенсивных параметров глубины реакции:
 . (II‑114)
. (II‑114)
Пример II‑3.
В раствор, в котором активность Ca2+ 10-3,5 (моль/л) и активности  10-1,5 (моль/л), поместили кусочек гипса. Надо определить, что будет с гипсом в стандартных условиях.
10-1,5 (моль/л), поместили кусочек гипса. Надо определить, что будет с гипсом в стандартных условиях.
Реакция растворения гипса имеет вид:

Если самопроизвольная реакция идет справа на лево, то гипс образуется, если слева на право, то он растворяется.
Чтобы определить направленность реакции необходимо сопоставить две величины: произведение активностей ее компонентов в растворе и стандартную константу равновесия. При столь низкой минерализации коэффициенты активности можно принять равными 1. Тогда произведение активностей для данной задачи можно определить как произведение концентраций по уравнению:
 .
.
Для очень разбавленных растворов активность растворителя, H2O, равна 1. Активность гипса, как чистого вещества, так же равна 1. Поэтому:
 =10-3,5×10-1,5 =10-5,0.
=10-3,5×10-1,5 =10-5,0.
Для определения константы равновесия воспользуемся величинами стандартных потенциалов свободной энтальпии из справочника. Согласно этим данным величины  имеют следующие значения в ккал/моль: CaSO4×2H2O -429,36; Ca2+ -132,35;
имеют следующие значения в ккал/моль: CaSO4×2H2O -429,36; Ca2+ -132,35;  -177,34; H2O -56,69. Отсюда следует:
-177,34; H2O -56,69. Отсюда следует:
= −132,35 −177,34−2 ×56,69 − (−429,36) = +6,29 (ккал/моль)
Тогда имеем
lgK0=-0,733×=-4,61; K0 =2,45×10-5=10-4,61,
Пa/Ka=10-5/10-4,61=10-0,39 , lg(Пa/Ka)=-0,39, DZ =RTln(Пa/Ka)=-0,53.
Это означает, что гипс в рассматриваемых условиях растворяется. При этом до достижения равновесия с гипсом в осадке раствору не хватает 0,53 ккал/моль полезной работы.
Скорость реакций
Одной из важнейших задач гидрогеохимии является определение изменения состава подземных вод во времени, для чего необходимо иметь представление о скоростях гидрохимических процессов в геологических условиях.
В экспериментальных работах под средней скоростью отдельных химических реакций в растворе обычно понимают вызванные ими изменения содержания вещества за единицу времени:
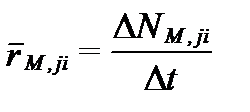 , (II‑115)
, (II‑115)
где 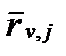 - средняя скорость реакции (mean reaction velocity)за время Δt, если число моль вещества за это время изменилась на ΔNM,ji. Скорость реакций не постоянна во времени и в действительности равна производной от изменения содержания по времени:
- средняя скорость реакции (mean reaction velocity)за время Δt, если число моль вещества за это время изменилась на ΔNM,ji. Скорость реакций не постоянна во времени и в действительности равна производной от изменения содержания по времени:
 . (II‑116)
. (II‑116)
Однако, например, в реакции фосфата кальция с серной кислотой:
Ca3(PO4)2 + 3SO4- +4H+ → 3CaSO4 + 2H2PO4-,
концентраций компонентов имеют разные скорости изменения. За один и тот же интервал времени 3 моля серной кислоты замещаются 2 молями ортофосфорной кислоты. В связи с этим, очевидно, что при оценке общей скорости реакции её необходимо нормировать по величине стехиометрических коэффициентов:

В теоретических работах такую скорость, равную производной от глубины химической реакции ξj по времени, называют истинной скоростью реакции (real reaction velocity).
Величина rj фактически характеризует расход потока веществ между реагентами и продуктами отдельной реакции j (моль·с-1) отнесенный к одному молю наименее затратного компонента. Однако, в практике химических исследований, как правило, имеют дело с концентрациями вещества в растворе. Поэтому используют истинные скорости реакции r,j нормированные по объему
:
 , (II‑117)
, (II‑117)
где V-объем раствора. Эта истинная скорость реакции нормированная по объему (reaction velocity normalized by volume)раствора используется, когда поток массообмена хаотичен, т.е. не имеет единой пространственной ориентации. В случае реакции между разными фазами (гетерогенные процессы), такой поток ориентирован относительно границы раздела. В этих случаях используют скорость реакции нормированную по площади (reaction velocity normalized by volume)поверхности разделаS, которую определяют по уравнению:
 . (II‑118)
. (II‑118)
В связи с этим справедливо равенство
 , (II‑119)
, (II‑119)
в котором rv,j и rs,j представляют собой удельные или нормированные скорости реакций (normalized reaction velocity)вмоль·м-3·с-1 и моль·м-2·с-1, соответственно.
Элементарные реакции
Большинство химических реакций представляет собой последовательную цепочку самых простых действий, которые называются актами или шагами. Такими шагами служат либо одновременное объединение двух, редко трех частиц (атомов, ионов, молекул, радикалов и т.д.) в одно соединение, либо, напротив, распад одного соединения на несколько. Реакции из одного акта называют элементарными реакциями (elementary reaction), а из нескольких актов – сложными (complex reaction).
Прежде всего, все элементарные реакции следует разделить на прямые и обратные. Прямыми реакциями называют те, в которых продукты образуются за счет реагентов, а обратными, наоборот, в которых реагенты образуются за счет продуктов. Прямые реакции в их уравнениях всегда идут слева направо, а обратные - справа налево. Поэтому скорости реакции не могут быть отрицательными: интервал времени всегда больше 0, а величины dCi или dξi могут быть отрицательными только у реагентов, у которых и стехиометрические коэффициенты отрицательные.
Элементарные химические реакции совершаются независимо друг от друга, но их скорость, последовательность и направленность определяют скорость и направленность всего химического процесса в целом. Скорости элементарных реакций зависят от количества и концентрации реагентов и от температуры.
Количество и концентрации реагентов. Скорость каждой элементарной реакции зависит, прежде всего, от количества одновременно участвующих в ней частиц, т.е. компонентов. Это минимальное число компонентов определяет молекулярность элементарной реакции, и выражается целым положительным числом: 1, 2 и очень редко 3. Вероятность столкновения 4 и более частиц одновременно ничтожно мала. Примером мономолекулярной элементарной реакции может служить диссоциация:
H2CO3 - → H++HCO3-.
Примером бимолекулятной элементарной реакции является ассоциация:
H++HCO3- → H2CO3.
Как отмечалось выше, норвежцы К.М. Гульдберг и П. Вааге в 1864 и 1867 гг. показали, что скорости таких реакций пропорциональны произведению концентраций их реагентов. Это открытие легло в основу главного постулата химической кинетики: скорость химических реакций пропорциональна произведению концентраций реагентов в степени их стехиометрических коэффициентов. Поэтому уравнение, которое связывает скорость реакции с концентрациями (или парциальными давлениями) реагентов имеет вид:
 , (II‑120)
, (II‑120)
и называется кинетическим уравнением (kinetic equation)или законом скорости реакции (reaction velocity law)реакции j. Здесь  - скорость реакции нормированная по объему, CM,ji –молярные концентрации реагентов, kr,j – некоторая постоянная величина, которая не зависит от содержания реагентов и называется константой скорости реакции (reaction velocity constant)или собственной скоростью реакции (intrinsic reaction velocity), а νji – стехиометрический коэффициент компонента i, называемый обычно парциальным порядком реакции (partial order of reaction). Сумма этих парциальных порядков одной реакции определяет порядок реакции (order of reaction)в целом или порядок её закона скорости (order of velocity law). Преобладают элементарные реакции (акты), подчиняющиеся закону скорости нулевого, первого и второго порядков. Например, для элементарной прямой реакции:
- скорость реакции нормированная по объему, CM,ji –молярные концентрации реагентов, kr,j – некоторая постоянная величина, которая не зависит от содержания реагентов и называется константой скорости реакции (reaction velocity constant)или собственной скоростью реакции (intrinsic reaction velocity), а νji – стехиометрический коэффициент компонента i, называемый обычно парциальным порядком реакции (partial order of reaction). Сумма этих парциальных порядков одной реакции определяет порядок реакции (order of reaction)в целом или порядок её закона скорости (order of velocity law). Преобладают элементарные реакции (акты), подчиняющиеся закону скорости нулевого, первого и второго порядков. Например, для элементарной прямой реакции:
2NO+ O2→ продукты
имеем:  ,
,
где парциальный порядок NO равен 2, и порядок реакции в целом - 3.
Нулевой порядок (zero-order reactions) означает отсутствие зависимости скорости реакции от концентрации реагентов. В таких реакцияхпри постоянной температуре скорость реакции неизменна во времени:
 , (II‑121)
, (II‑121)
и kr,j имеет размерность скорости реакции (моль·м-3·с-1). Содержание реагента при таких реакциях уменьшается линейно, согласно уравнению:
CM,t,ji=CM,0,ji – kr,j·Δt, (II‑122)
где CM,0,ji- начальное содержание компонента i. Время уменьшения содержания реагента вдвое называется периодом полураспада (half-life period) . При скорости нулевого порядка этот период равен:
 . (II‑123)
. (II‑123)
Первый порядок (first-order reaction) означает прямую линейную зависимость скорости реакции от содержания реагента. Реакции первого порядка имеют вид:
Ci → продукты.
Например, в реакции
H2S → HS- + H+
формула скорости имеет вид
 .
.
Скорость такой реакции определяется общим уравнением
 . (II‑124)
. (II‑124)
где kr,j имеет размерность (с-1). Интегрируя это уравнение в интервале от начального содержания реагента CM,0,ji до CM,t,ji , получим:
 . (II‑125)
. (II‑125)
Отсюда следует, что содержание реагента i в любой момент времени t зависит только от его начального содержания и продолжительности реакции, согласно уравнениям
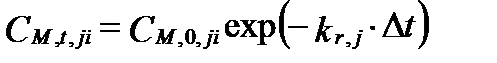 или lnCM,t,ji= lnCM,0,ji – kr,jΔt . (II‑126)
или lnCM,t,ji= lnCM,0,ji – kr,jΔt . (II‑126)
К реакциям первого порядка относится реакции диссоциации ионов, многие реакции разложения органических соединений и радиоактивный распад. При этих реакциях величина периода полураспадаопределяется относительно простым уравнением:
 . (II‑127)
. (II‑127)
Отсюда следует, что произведение константы скорости на период полураспада (τj·krj) в реакциях первого порядка всегда равно 0,693.
Второй порядок (second-order reaction)свойственен бимолекулярным элементарным реакциям, которые имеют вид:
C1+ C2→ продукты.
Скорость этих реакций определяется уравнением:
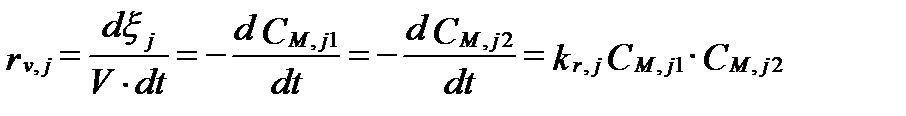 , (II‑128)
, (II‑128)
в котором kr,j имеет размерность (моль-1·м3·с-1). Примером может служить реакция:
Fe3+ + SO42-→ FeSO4+,
формула скорости которой имеет вид
 .
.
В частном случае, когда С1 = С2имеем
 . (II‑129)
. (II‑129)
Интегрируя это уравнение в интервале от начального содержания CM,0,ji до CM,t,ji получим
 . (II‑130)
. (II‑130)
Отсюда следует, что содержание реагента i меняется со временем согласно уравнению
 . (II‑131)
. (II‑131)
Если известна величина потери ∆CM,t,ji= CM,0,ji – CM,t,ji, то
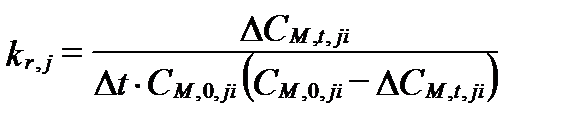 . (II‑132)
. (II‑132)
Поэтому при ∆CM,t,ji= 0,5CM,0,ji и равенстве начальных концентраций период полураспада равен
 . (II‑133)
. (II‑133)
В другом частном случае один из реагентов присутствует в таком большом количестве, что его относительные расходы при реакции второго порядка ничтожны. Тогда реакция второго порядка ведет себя, как реакции первого порядка. Напрмер, если в реакции участвует сам растворитель H2O, изменение его относительного содержания ничтожно мало и практически не влияет на изменение скорости реакции. Тогда
 . (II‑134)
. (II‑134)
Такие реакции называются псевдомономолекулярными (pseudo-monomolecular)или псевдоперврго порядка.
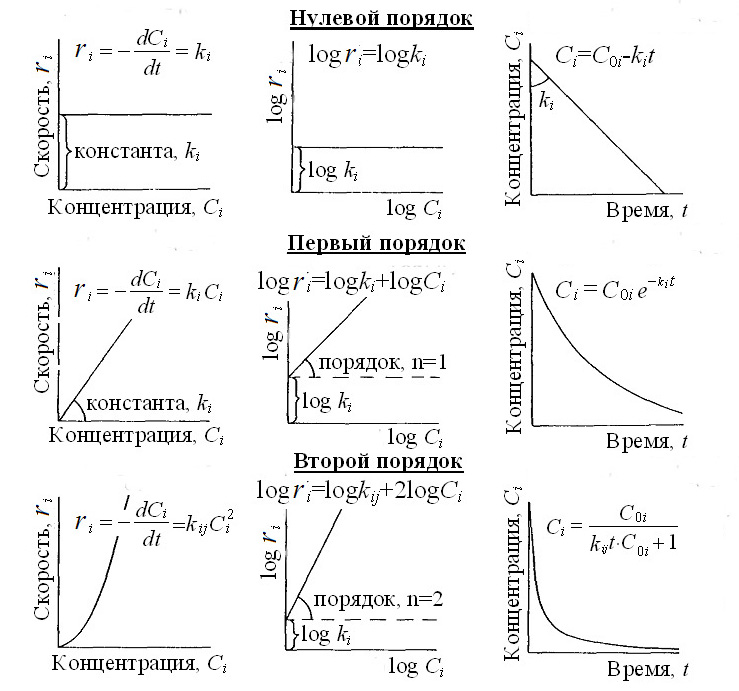
|
| Рисунок II-13. Уравнения и графики зависимостей реакций разного порядка. |
Температура. Во всех рассмотренных уравнениях константы скорости реакций kr,j численно равны самой скорости при Cji равных единице. Но её размерность зависит от порядка закона скорости и равна с-1·(моль·м-3)1-η, где η – порядок реакции. Эти константы характеризуют зависимость скорости реакции от энергетического потенциала взаимодействующих компонентов. По своей сути элементарная прямая реакция это результат перехода части кинетической энергии движения отдельных реагентов в потенциальную энергию продуктов реакции. Таким образом, кинетическая энергия движения противостоит потенциальной энергии межмолекулярных и межатомных связей. Чтоб акт реакции совершился, необходима такая кинетическая энергия движения в растворе, которая способна преодолеть силы отталкивания при сближении, разрушить гидратную оболочку соединения, а затем и связи внутри компонента. Таким образом, реагенты при столкновении должны преодолеть некоторый энергетический барьер, величина которого зависит от природы реагентов. Гипотезу, что элементарная реакция совершается только при наличии достаточной кинетической энергией, предложил Сванте-Август Аррениус (1859- 1927), который в 1889 году обнаружил зависимость величины константы скорости реакции от температуры. Эта зависимость имела вид:
 , (II‑135)
, (II‑135)
где A и B – константы, характерные для данной реакции, а T – абсолютная температура. С. Аррениус предположил, что каждый элементарный акт реакции имеет две стадии:
A+B↔A*+B* → продукты.
На первой стадии реагенты сталкиваются и переходят в энергетически возбужденное, активированное состояние A* и B*. На этой стадии реакция еще обратима. На второй стадии некоторые активированные реагенты взаимодействуют и образуют продукты реакции. На этом основании он предложил уравнение
 , (II‑136)
, (II‑136)
в котором ke,j – предэкспоненциальный коэффициент, а Ea,j - энергия активации (activation energy)реакции j. Уравнения II-129 и II-130 называются уравнениями Аррениуса (Arrhenius equation). Величины предэкспоненциального коэффициентаke,j и энергии активации Ea,j обычно определяют опытным путем по температурной зависимости константы скорости реакции. Из последнего уравнения видно, что зависимость константы скорости реакции и самой скорости реакции от температуры тем больше, чем выше энергия активации.
Таким образом, скорость элементарной реакции и её поведение зависят от числа и концентрации реагентов и температуры. Эта зависимость может быть выражена общим уравнением:
 , (II‑137)
, (II‑137)
Где предэкспоненциальный коэффициент ke,j имеет размерность константы скорости.
Пример II‑4.
Определить скорость окисления Fe2+в нормальных условиях при величинах pH 5 и 7, если его концентрация  = 1 ммоль/л, парциальное давление кислорода
= 1 ммоль/л, парциальное давление кислорода 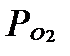 = 0,2 атм, а константа скорости по железу
= 0,2 атм, а константа скорости по железу  , согласно данным Stumm W., Morgan J.J. (1981), равна 8,0 1013 мин-1. атм-1.
, согласно данным Stumm W., Morgan J.J. (1981), равна 8,0 1013 мин-1. атм-1.
Реакция окисления:
Fe2++O2+2OH-→Fe3++2H2O
Скорость окисления Fe2+ в водных растворах при 20°C и 1 атм, в соответствии с приведенным уравнением скорости:

Тогда при pH= 5  =10-14-10-5=10-9, а
=10-14-10-5=10-9, а  =8,0×1013∙10-3∙(10-9)2∙ 0,2= l,6∙10-8 моль×л-1×мин-1 =9,6×10-4ммоль×л-1час-1, а при pH= 7 =10-14-10-7=10-7, а
=8,0×1013∙10-3∙(10-9)2∙ 0,2= l,6∙10-8 моль×л-1×мин-1 =9,6×10-4ммоль×л-1час-1, а при pH= 7 =10-14-10-7=10-7, а  =8,0×1013∙10-3 ∙ (10-7)2∙ 0,2= 1,6∙10-4 моль×л-1×мин-1 = 9,6 ммоль×л-1час-1. Обращает на себя внимание резкое увеличение скорости реакции при изменении pH всего на две позиции. Это обусловлено не только тем, что этим двум позициям соответствует увеличение концентрации OH- в 100 раз, но и вторым порядком зависимости скорости -. При удвоении концентрации
=8,0×1013∙10-3 ∙ (10-7)2∙ 0,2= 1,6∙10-4 моль×л-1×мин-1 = 9,6 ммоль×л-1час-1. Обращает на себя внимание резкое увеличение скорости реакции при изменении pH всего на две позиции. Это обусловлено не только тем, что этим двум позициям соответствует увеличение концентрации OH- в 100 раз, но и вторым порядком зависимости скорости -. При удвоении концентрации  или
или 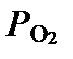 скорость реакции увеличивается в два раза, тогда как при таком же увеличении концентрации OH- скорость растет в 4 раза.
скорость реакции увеличивается в два раза, тогда как при таком же увеличении концентрации OH- скорость растет в 4 раза.
Сложные реакции
Большинство химических реакций в природе содержит несколько связанных между собой моно- или бимолекулярных шагов, и поэтому имеет сложный, часто обратимый характер. Всю последовательность и взаимоотношения элементарных актов сложной реакции называют механизмом (reaction mechanism). Механизмы сложных реакций различаются не только количеством и свойствами элементарных актов, но и характером их последовательности и направленности. Наиболее распространенными в гидрохимии являются последовательные, параллельные, сопряженные и обратимые механизмы.
Механизм, при котором реагент A прежде, чем превратиться в D, образует промежуточные временные продукты B и C согласно уравнению:

называется последовательным. В таких реакциях ведущую роль играют самые медленные химические акты, которые фактически и определяют закон их общей скорости. Содержание промежуточных продуктов в этих реакциях определяется соотношением скоростей их образования и удаления. Если у промежуточного продукта скорость удаления значительно больше скорости образования, может сформироваться стациионарный режим, при котором общая скорость реакции стабилизируется.
Параллельными называют реакции, которые включают два или более последовательных механизмов на пути от реагента A до продукта P.

При этом механизме определяющую роль играет та последовательность, которая обладает наибольшей скоростью. Если параллельные реакции имеют заметно разные скорости, то та, что имеет большую скорость, обычно называют главной, а остальные – побочными.
Многие сложные реакции обратимы, т.е. могут идти в двух противоположных направлениях. В этом случае общую скорость реакции определяют, как разность между скоростями этих противоположно направленных реакций:
 , (II‑138)
, (II‑138)
где  и
и  скорости прямой и обратной реакций, соответственно.
скорости прямой и обратной реакций, соответственно.
Рассмотрим одну из самых простых и наиболее распространенных сложных реакций, а именно обратимую реакцию первого порядка:
A↔B.
В этой реакции имеются две встречные элементарные реакции, скорости которых равны
 и
и  (II‑139)
(II‑139)
Общая скорость такой реакции будет равна разности скоростей в прямом и обратном направлении:
 (II‑140)
(II‑140)
Эта общая скорость в закрытой системе будет уменьшаться до тех пор, пока скорости элементарных актов не уравняются, т.е. пока не будут достигнуты равенства:
 или
или  .
.
Из них следует, что в случае химического равновесия
 . (II‑141)
. (II‑141)
С другой стороны, химическое сродство такой реакции, согласно уравнению II-104 равно:
 . (II‑142)
. (II‑142)
Если принять соотношение коэффициентов активности γjB/γjA равным 1, и выразить константу равновесия через соотношение скоростей реакции в уравнении II-135, то получим:
 (II‑143)
(II‑143)
Из последнего уравнения следует, что
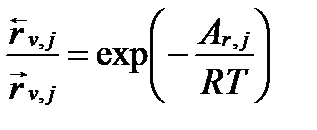 . (II‑144)
. (II‑144)
Подставим это выражение в уравнение II-134 и получим уравнение общей скорости обратимой реакции первого порядка:
 (II‑145)
(II‑145)
Это уравнение показывает, что скорости обратимых реакций зависият не только от концентраций реагентов, но и от величины химического сродства Ar,j или степени насыщения Ωj. В условиях равновесия, когда  , величина Ar,j = 0, а Ωj = 1, суммарная скорость обратимой реакции равна 0. По мере роста недоонасыщенности продуктами величина Ωj опускается ниже 1, когда величина Ar,j поднимается выше 0, и общая скорость реакции растет до некоторого предела равного
, величина Ar,j = 0, а Ωj = 1, суммарная скорость обратимой реакции равна 0. По мере роста недоонасыщенности продуктами величина Ωj опускается ниже 1, когда величина Ar,j поднимается выше 0, и общая скорость реакции растет до некоторого предела равного  . В связи с этим выражение в квадратных скобках удообно включить в константу скорости обратимой реакции:
. В связи с этим выражение в квадратных скобках удообно включить в константу скорости обратимой реакции:
 . (II‑146)
. (II‑146)
Природные химические процессы обычно слишком сложны, чтобы механизм их сложных химических реакций был однозначно определен, а констант их скоростей рассчитаны. В большинстве гидрохимиических процессов константы скоростей реакций и их зависимость от термодинамических параметров определяются экспериментально. При этом формально допускается, что константы скоростей сложных реакций подчиняются тем же законам, что и константы скоростей элементарных реакций.
Прежде всего, полагают, что суммарная скорость сложной реакции так же подчинена главному постулату химической кинетики, и её величина определяется уравнением:
 . (II‑147)
. (II‑147)
В практике экспериментальных исследований эту зависимость определяют, как функцию изменения концентрации одного или двух реагентов. Тогда скорость даже очень сложных реакций описывается относительно простым уравнением типа:
 . (II‑148)
. (II‑148)
В этом случае нормирующий стехиометрический коэффициент компонента i, как и влияние остальных соединений реакции, включается в константу скорости kr,j. Компонентами, контролирующими скорость реакций, обычно являются H+ или OH-, CO2, O2, Fe3+ и т.д. Значения их парциального порядка νji могут отличаться от величин их стехиометрических коэффициентов в уравнениях реакций, и быть дробным или даже отрицательным числом. Это связано с тем, что один и тот же компонент может участвовать в разных элементарных актах одного механизма сложной реакции.
Константа скорости kr,j в последнем уравнении численно равна скорости реакции, когда концентрация CM,j равны 1 молю на литр при размерности с-1·(моль·м-3)1-η, где η – порядок реакции. Она включает в себя влияние всех остальных факторов, прежде всего, температуры T и степени насыщения Ωj.
Влияние температуры зависит от энергии активации процесса и определяется уравнением Аррениуса. Оно может быть включено в константу скорости сложных реакций по аналогии с уравнением II-130.
Влияние степени насыщения связано с обратимостью сложных реакций. Направленность таких природных реакций связана с относительным содержанием их рагентов и продуктов. Их суммарная скорость падает по мере уменьшения концентрации реагентов и роста содержания продуктов. Поэтому с приближением величины степени насыщения Ωj к 1, или химического сродства Ar,j к 0 константа суммарной скорости стремится к 0.
Если допустить принцип микроскопической обратимости), согласно которому прямые и обратные механизмы только направленностью одинаковых элементарных актов, то влияние температуры, степени насыщения или химического сродства на константу скорости обратимых реакций можно объединить в одном уравнении:
 . (II‑149)
. (II‑149)
Здесь p и q – эмпирические безразмерные параметры, которые учитывают возможное отклонение от принципа микроскопической обратимости). Тогда общее уравнение суммарной скорости сложной химической реакции можно представить в виде:
 (II‑150)
(II‑150)
Единица в квадратных скобках этого уравнения характеризует отношение величин обратной и прямой скорости, когда они равны друг другу, а второе слагаемое в этих скобках определяет фактическое соотношение тех же скоростей в растворе. С уменьшением разности между этими слагаемыми величина суммарной скорости реакции стремится к 0. Напротив, с приближением величины второго слогаемого к 0, разность в квадратных скобках приближается к 1. В этом случае скорость сложной реакции стремится к некоторому пределу, который зависит только от концентрации реагентов и температуры. По своей сущности этот предел представляет собой величину только прямой скорости реакции, т.е.  , которую часто именуют начальной, максимальной или далекой от равновесия (initial, maximal, reaction velocity far from equilibrium). Тогда суммарную скорость сложной реакции можно представить в виде:
, которую часто именуют начальной, максимальной или далекой от равновесия (initial, maximal, reaction velocity far from equilibrium). Тогда суммарную скорость сложной реакции можно представить в виде:
 . (II‑151)
. (II‑151)
Таким образом, скорости многих сложных реакций в природных условиях можно выразить как функцию их максимальной прямой скорости 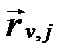 , полученной экспериментально в лабораторных условиях для идеальных разбавленных растворов, и величин их степени насыщения Ωj или химического сродства
, полученной экспериментально в лабораторных условиях для идеальных разбавленных растворов, и величин их степени насыщения Ωj или химического сродства 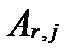 .
.
Величины этих максимальных скоростей реакций в природных водах меняются в очень широком диапазоне. Обычно их сравнивают по величине полураспада.
Химический потенциал
Обмен электронами
Многие растворенные элементы способны менять свою валентность и иметь в растворе сразу несколько степеней окисления. Это связано с тем, что электроны, стабильные элементарные частицы e- с массой 9,1·10-31 кг и электрическим зарядом -1, способны в составе раствора переходить от одного атома к другому и менять их степень окисления и свойства. Такие реакции обмена атомов электронами с изменением их степени окисления называются окислительно-восстановительными. Под окислением понимают потерю электронов, а под восстановлением – их приобретение. Поэтому, компоненты-донары, способные отдать электроны, называют восстановителями (reducer), а атомы-акцепторы, способные их принять, окислителями (oxidizer). Реакции окисления и восстановления всегда связаны между собой. Любую окислительно-восстановительную реакцию, например:
O2+4Fe2++4H+= 2H2O +4Fe3+,
можно представить в виде двух полуреакций:
окисления 4Fe2+ = 4Fe3+ + 4e-;
восстановления O2 +4H+ + 4e- = 2H2O.
Такие окислительно-восстановительные реакции часто называют редокс-реакциями, а сопряженные ими компоненты - редокс парами (redox couple, redoxpara).
Рисунок II-14. Наиболее важные элементы с переменной степенью окисления в природе.
| Элементы | степени окисления | Примеры соединений | Элементы | степени окисления | Примеры соединений |
| O | O2 | Fe | +3 | Fe3+, Fe(OH)3 | |
| -1 | H2O2 | +2 | Fe2+ | ||
| -2 | H2O, CO2 | Mn | +4 | MnO2, MnO42- | |
| C | +4 | CO2,  , , 
| +3 | Mn(OH)3, Mn3+ | |
| CH2O, C | +2 | Mn2+ | |||
| -4 | CH4, CH3OH, C6H6 | U | +6 | UO22+, UO2H+ | |
| S | +6 | 
| +4 | U(OH)3+, U4+ | |
| +5 | 
| ||||
| +2 | 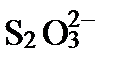
| ||||
| -2 | H2S, HS- | ||||
| N | +5 | 
| |||
| +3 | 
| ||||
| N2 | |||||
| -3 | NH3, 
| ||||
| P | +5 | PO43-, PO3- | |||
| +3 | PO33- |
Многие элементы в составе природных вод способны менять степень окисления и поэтому являются либо окислителями, либо восстановителями. Наиболее распространенные и важные из них представлены в таблице II-7. Наиболее характерные полуреакции восстановления и их константы равновесия представлены в таблице II-8. Во всех этих полуреакциях участвуют электроны e-. Относительное количество растворенных окислителей и восстановителей определяет содержание электронов, способных к обмену между элементами раствора, и тем самым определяет общие окислительно-восстановительные свойства природных вод.
Рисунок II-15. Восстановительные полуреакции природных вод.
| Элементы | Полуреакции восстановления | pe0= 
| E0, В | |||||||
| H+ | 2H++ 2e-= H2 (г)
Популярное: Как выбрать специалиста по управлению гостиницей: Понятно, что управление гостиницей невозможно без специальных знаний. Соответственно, важна квалификация... Как распознать напряжение: Говоря о мышечном напряжении, мы в первую очередь имеем в виду мускулы, прикрепленные к костям ... Как построить свою речь (словесное оформление):
При подготовке публичного выступления перед оратором возникает вопрос, как лучше словесно оформить свою... Генезис конфликтологии как науки в древней Греции: Для уяснения предыстории конфликтологии существенное значение имеет обращение к античной...  ©2015-2024 megaobuchalka.ru Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. (1216)
|
Почему 1285321 студент выбрали МегаОбучалку... Система поиска информации Мобильная версия сайта Удобная навигация Нет шокирующей рекламы |
(0.016 сек.)