 |
Главная |


Порядок сбора генеалогической информации и методика составления родословной
|
из
5.00
|
Начинать сбор сведений о родословной следует от пробанда, то есть человека (больного, здорового), обратившегося за консультацией. Сбор генеалогической информации о наличии среди его родственников различных заболеваний может проводиться разными методами: опросом, анкетированием, личным обследованием членов семьи.
В генетическую карту подробно записываются все сведения о пробанде (анамнез настоящего заболевания, его начало, последующее течение, возраст, в котором появились первые признаки заболевания). В дальнейшем собирают данные об его сибсах (братьях и сестрах) и родителях. Далее опрос продолжается о родственниках по линии матери. Запись сведений удобнее вести в следующем порядке: родители матери, их дети по порядку рождения с указанием потомков (внуков). Очень важно выяснить у женщин, как протекала беременность, на каком фоне она наступила, подробности о всех случаях выкидышей, мертворождений, о наличии бесплодных браков и ранней детской смертности. В такой же последовательности собираются сведения о родственниках отца пробанда. Естественно, чем больше родственников пробанда будет непосредственно опрошено или обследовано, тем выше шансы на получение более достоверных и полезных сведений.
При сборе данных о родословной необходимо отмечать девичьи фамилии женщин и место жительства семьи. Это помогает выявить кровно-родственные браки, которые влияют на появление наследственных болезней и их частоту. Если родители пробанда проживали в одном небольшом по числу жителей районе (особенно изолированном географически), можно предположить, что они имеют общих предков, а, следовательно, и большее количество общих патологических генов.
При составлении родословной очень важно учесть наличие и характер профессиональных вредностей (особенно для родителей, имеющих детей с врожденными пороками развития), время их действия (до беременности или во время нее). Следует также выявить факторы, которые могли повлиять на возникновение эмбриопатии (прием лекарственных препаратов в первые недели беременности, заболевания матери в данный период, рентгеновское облучение женщины и др.).
ГРАФИЧЕСКОЕ ИЗОБРАЖЕНИЕ РОДОСЛОВНОЙ
После сбора генеалогической информации переходят к графическому изображению родословной. Наиболее распространенные символы, используемые при ее составлении, представлены на рис. 26.
При графическом изображении родословной необходимо соблюдать определенные правила.
1. Составление родословной начинают с пробанда. Братья и сестры располагаются в родословной в порядке рождения слева направо, начиная со старшего.
2. Все члены родословной должны располагаться строго по поколениям в один ряд.
3. Поколения обозначаются римскими цифрами слева от родословной сверху вниз.
4. Арабскими цифрами нумеруется потомство одного поколения (весь ряд) слева направо последовательно. Таким образом, каждый член родословной имеет свой шифр (например: I-3, II-2 и т. д.).
5. Необходимо указывать возраст членов семьи, так как некоторые наследственные заболевания проявляются в разные периоды жизни.
6. Супруги родственников пробанда могут не изображаться в родословной, если они здоровы.
7. Важно отметить лично обследованных членов родословной знаком (!).
Составляя родословную, желательно получить сведения о максимальном количестве родственников 3-4поколений. Если рассматриваемых признаков несколько, то можно прибегать к буквенным или штриховым различиям внутри символов (рис. 18).

Рис. 18. Родословная семьи с сахарным диабетом (обозначено штриховкой) и с синдромом Марфана (обозначено чёрным цветом)
Все полученные данные о состоянии здоровья родственников, причина и возрасте смерти записываются внизу под родословной (легенда) с указание даты ее составления. Тщательно собрав все данные, можно приступить к анализу родословной. Рассмотрим основные типы наследования моногенных болезней.
АУТОСОМНО-ДОМИНАНТНЫЙ ТИП НАСЛЕДОВАНИЯ
При данном типе наследования наиболее часто встречаются браки между больными (Аа) и здоровыми членами семьи (аа), где А - доминантный ген, определяющий развитие наследственного заболевания, а - рецессивный ген.
Для аутосомно-доминантного типа наследования характерны следующие признаки:
1) передача заболевания из поколения в поколение (наследование по вертикали);
2) передача заболевания от больных родителей детям;
3) здоровые члены семьи обычно имеют здоровое потомство;
4) оба пола поражаются одинаково часто (рис. 28).
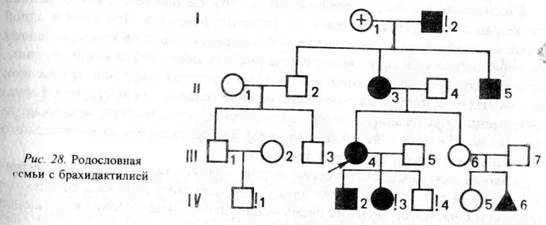
Пробанд (IП-4) унаследовала дефект от своей матери (II-3) и деда (I-2) и передала его своим детям (IV-2, 3).
По аутосомно-доминантному типу наследуются синдром Марфана, поли- дактилия, брахидактилия, синдактилия, ахондроплазия, анемия Минковского- Шоффара и многие другие заболевания.
В случае доминантного типа наследования, если один из родителей болен (Аа), прогноз потомства может быть определен еще до рождения ребенка. Он равен 50 % при условии гетерозиготности больного и полной пенетрантности гена.
Если ген встречается в популяции часто, то при аутосомно-доминантном типе наследования возможны браки типа: АахАа, в которых могут появиться дети гомозиготы по данному заболеванию. В таких семьях вероятность рождения больного ребенка равна 75 %. Однако многие аутосомно-доминантные заболевания в гомозиготном состоянии протекают значительно тяжелее, чем у гетерозигот, и дают, как правило, летальный исход.
Иногда при анализе родословной возникает следующая ситуация: пробанд страдает доминантной болезнью (например, отосклерозом), его мать здорова, а бабушка также страдала той же болезнью. В этом случае мать пробанда - носитель патологического гена, так как она передала его своему ребенку, но у неё он не проявился, то есть не пенетрировал. В результате вид родословной изменяется и появляются пропуски поколений.
Пенетрантность - это частота проявления гена среди носителей данного гена. Она представляет собой отношение особей, имеющих данный признак, к особям, имеющим данный ген, выраженное в процентах. Так, пенетрантность отосклероза - 40 %, синдрома Марфана - 30 %, ретинобластомы - 80 %.
Собирая анамнез и анализируя родословную, нельзя забывать и о другом свойстве гена - его различной экспрессивности. Экспрессивность - степень выраженности гена. Понятие экспрессивности аналогично понятию тяжести заболевания. При высокой экспрессивности гена развивается тяжёлая форма заболевания, часто с летальным исходом, при низкой - создаётся впечатление, что человек здоров.
Рассмотрим понятие экспрессивности гена на примере наследственной патологии соединительной ткани - синдроме Марфана. При этом в одной семье можно встретить как очень тяжелые формы с классическим поражением костной системы (в виде сколиоза или кифосколиоза, деформации грудины, арахнодактилии); нарушением зрения (двусторонний подвывих хрусталика) и сердечно-сосудистой системы (расширение аорты), так и стертые формы заболевания, протекающие легко (астеническое телосложение, арахнодактилия, сколиоз I степени, небольшая миопия). Таких людей без дополнительного обследования можно отнести к здоровым.
Слабовыраженные клинические формы болезни могут легко просматриваться, тогда родословная также теряет свой «классический» вид, появляются пропуски поколении. Поэтому очень важно личное обследование всех членов семьи.
АУТОСОМНО-РЕЦЕССИВНЫЙ ТИП НАСЛЕДОВАНИЯ
Для данного типа наследования характерны следующие признаки: а) больные дети рождаются от фенотипически здоровых родителей, являющихся гетерозиготными носителями патологического гена.
Генотипы родителей
Мать Аа Отец Аа
Генотипы детей
АА Аа, Аа аа
(здоровый) (здоровые, но носи- (больной)
тели аномального гена)
Как видно из схемы, болеют только гомозиготы по рецессивному гену;
б) больные чаще встречаются в одном поколении: среди родных или двоюродных сибсов (наследование «по горизонтали») или среди дядей и племянников (наследование «по ходу шахматного коня»);
в) в родословной отмечается более высокий процент кровно-родственных браков;
г) одинаково часто болеют мужчины и женщины (рис. 19).
К заболеваниям с аутосомно-рецессивным типом наследования относятся фенилкетонурия, галактоземия, альбинизм, муковисцидоз, целиакия, большинство мукополисахаридозов и др.
Рассмотрим родословную семьи с альбинизмом (см. рис. 19).

Рис. 19. Родословная семьи с альбинизмом
Альбинизм - редкая аутосомно-рецессивная патология аминокислотного обмена, при которой страдает синтез пигмента меланина. Альбиносы имеют белую кожу, белые волосы, обесцвеченную радужную оболочку глаз (красные глаза). Кожа и глаза очень чувствительны к солнечному свету. Как видно из рис. 19, больные чаще встречаются в одном (IV) поколении среди родных и двоюродных сибсов; в III поколении родословной имеются родственные браки. Больные в данной семье появились только в III и IV поколениях. Это-особенность рецессивного гена существовать во многих поколениях в гетерозиготном состоянии, никак не проявляясь фенотипически. Поэтому первый больной рецессивной болезнью может появиться через многие поколения после возникновения мутации, так как рождение больного ребенка возможно только в том случае, если оба родителя являются гетерозиготами (Аа).
Риск рождения больного ребенка в такой семье равен 25 %.
Однако при аутосомно-рецессивном типе наследования редко, но возможны и другие варианты браков:
аа х аа - у таких родителей все дети будут больными (аа).
Аа х аа - 50 % детей будут больными (генотип аа), 50 % фенотипически
здоровыми (генотип Аа), но будут являться носителями мутантного гена.
Следует отметить, что частота рецессивных наследственных болезней особенно повышается в изолятах и среди населения с высоким процентом кровнородственных браков. Об отрицательном влиянии родственных браков на потомство свидетельствует и тот факт, что умственная отсталость среди детей от этих браков в 4 раза выше, чем в семьях с неродственными браками.
Х-СЦЕПЛЕННЫЙ ТИП НАСЛЕДОВАНИЯ
В Х-хромосоме могут локализоваться как доминантные, так и рецессивные гены. У человека известно более 200 генов, в основном патологическиx сцепленных с Х-хромосомой.
У женщины аномальный ген может находиться в одной (гетерозигота) или обеих (гомозигота) Х-хромосомах; у мужчин - только в одной Х-хромосоме, поэтому не только доминантный, но и рецессивный ген в его Х-хромосоме всегда будет проявляться. Такое состояние гена называется мизиготным.
Если в Х-хромосоме локализуется рецессивный ген, то такой тип наследования называется Х-сцепленным рецессивным. Для этого типа наследования характерны следующие признаки:
а) болеют преимущественно лица мужского пола;
б) больные дети рождаются от фенотипически здоровых родителей, ни мать больного является гетерозиготной носительницей патологического гена («кондуктор»);
в) больные мужчины не передают заболевания своим сыновьям, но их дочери становятся «кондукторами».
г) редкие случаи заболевания женщин возможны, если их отец болен, мать - носительница (рис. 30).
Если в брак вступает здоровый мужчина и гетерозиготная женщина, вероятность рождения больного мальчика в данной семье составляет 50% от всех мальчиков (или 25 % от всех детей), а все девочки этих родителей будут здоровы, но половина из них станут носительницами патологического гена.
В случае, если в брак вступают больной мужчина и здоровая женщина, все дети будут здоровыми, но дочери получат от отца мутантный аллель и станут «кондукторами».
Третий вариант браков, который встречается редко при рецессивном сцепленном с Х-хромосомой наследовании, - это брак между больным мужчиной и женщиной гетерозиготой. В этом случае ожидается рождение половины больных мальчиков; половина девочек также будет больна, а половина - станет носительницами патологического гена.
Классическим примером рецессивного сцепленного с Х-хромосомой наследования является гемофилия.
Клиника заболевания связана с тем, что из-за недостатка необходимых для свертывания крови факторов больные страдают кровоточивостью. Даже после небольших травм возникают значительные кровопотери, развиваются анемия, гемартрозы. На рис. 30 изображена родословная семьи с гемофилией А.

Пробанд (III - 1), его двоюродный брат (III - 8) и их дядя (II - 3) больны гемофилией. Бабка пробанда (I - 1), его мать (II - 2) и его тётка (II - 5) - носители рецессивного сцепленного с Х-хромосомой гена гемофилии А.
Кроме гемофилии, рецессивный сцепленный с Х-хромосомой тип наследования характерен для псевдогипертрофической миопатии Дюшенна, агаммаглобулинемии Брутона, некоторых форм дальтонизма и др.
Если в хромосоме X локализуется доминантный ген, такой тип наследования называется Х-сцепленным доминантным. Для него характерны следующие признаки:
а) заболевание прослеживается в каждом поколении;
б) если болен отец, то все его дочери будут больными, а все сыновы здоровыми;
в) если больна мать, то вероятность рождения больного ребенка равна 50 % независимо от пола;
г) болеют как мужчины, так и женщины, но в целом больных женщин в семье в 2 раза больше, чем больных мужчин;
д) у здоровых родителей все дети будут здоровыми.
Анализ родословной, изображенной на рис. 31, показывает, что дети с коричневыми зубами рождаются от браков, в которых один родитель болен а другой здоров. Все здоровые члены родословной имеют здоровое потомство. Обращает на себя внимание третье поколение, где все мужчины (III – 2, III -3) унаследовали нормальную окраску эмали зубов, а все женщины (III – 4,Ш-6, III - 7) унаследовали дефект окраски эмали. Их отец (II - 2) имел коричневые зубы, а мать (II - 1) - белые. Следовательно, ген коричневой окраски эмали зубов находится в Х-хромосоме. Только в этом случае он не мог оказаться у сыновей, но обязательно должен был попасть к дочерям. При этом он проявился у всех дочерей.

Сцепленное с Y-хромосомой наследование характеризуется тем, что гены, локализованные в Y-хромосоме, передаются только сыновьям пораженного отца, а его дочери остаются здоровыми, так как они никогда не получают Y-хромосомы от отца (голандрическое наследование). По такому типу у человека наследуются «мохнатые уши» - наличие волос по краю ушных раковин.
МУЛЬТИФАКТОРИАЛЬНОЕ НАСЛЕДОВАНИЕ
Мультифакториальные болезни, или болезни с наследственным предрасположением, составляют в настоящее время 92 % от общей патологии человека. Они вызываются изменением чаще нескольких генов и для своего проявления требуют влияния факторов внешней среды.
К наиболее часто встречающимся мультифакториальным заболеваниям относятся: ревматизм, ишемическая болезнь сердца, гипертоническая болезнь, эпилепсия, мигрень, язвенная болезнь, цирроз печени, неспецифический язвенный колит, сахарный диабет, бронхиальная астма, псориаз, шизофрения и др. При перечисленных болезнях из поколения в поколение передается предрасположенность к определенному заболеванию.
Мультифакториальные болезни при всем их разнообразии имеют некоторые общие черты:
1) высокая частота в популяции (например, сахарным диабетом страдает около 5 % населения промышленно развитых стран, аллергическими заболеваниями - более 10 %, шизофренией - 1 % населения, гипертонией около 30 %);
2) несоответствие наследования законам Менделя;
3) существование клинических форм от скрытых субклинических до резко выраженных проявлений;
4) более раннее начало заболевания и некоторое усиление клинических проявлений в нисходящих поколениях.
Генетический прогноз при мультифакториальном типе наследования зависит от следующих факторов:
а) частоты встречаемости заболевания в популяции (чем она ниже, тем выше риск возможности заболевания для родственников пробанда);
б) степени выраженности болезни у пробанда (чем она выше, тем больше риск развития болезни у родственников, так как тяжесть заболевания определяется суммарным эффектом более чем одного гена). Например: человек, получивший два гена, от которых зависит артериальная гипертензия, может иметь более тяжелую степень заболевания и в два раза большую вероятность передачи патологического гена потомству;
в) степени родства с пораженным членом семьи, так как это определяет число общих генов у данного человека с больным.
Наибольшее число общих генов (100 %) имеется у монозиготных близнецов. Различия в фенотипе у них могут быть вызваны только факторами внешней среды, поэтому близнецовый метод является ведущим в определении роли наследственных и средовых факторов в развитии мультифакториальных заболеваний.
Каждый родитель передает своему ребенку половину своего хромосомного набора, то есть половину генов, затем с каждым новым поколением у потомков число генов, одинаковых с каким-то общим предком, уменьшается вдвое. Чем больше общих генов, тем больше возможность однотипного влияния двух или более патологических генов, если они были у общего предка. Поэтому важно оценить степень родства с больным, то есть число общих генов (табл. 3).

При анализе родословных риск проявления заболевания велик при болезни родственников I и II степеней родства. При наличии болезни среди родственников III степени родства он может рассматриваться как умеренный. Единичные, спорадические случаи болезни среди родственников IV и более дальних степеней родства указывают на малую степень риска;
г) число больных родственников также определяет прогноз для пробанда.
Чем больше в родословной больных, тем выше риск проявления болезни. Так, при сахарном диабете риск для сибсов пробанда в зависимости от числа больных родственников будет следующим: 1) если родители здоровы, вероятность заболевания равна 5-10 %; 2) если болен один из родителей, риск увеличивается до 10-20 %; 3) если больны оба родителя, риск заболеть сахарным диабетом для сибсов пробанда будет равен 20-40 %;
д) в случаях разницы в частоте заболевания по полу риск для родственников будет выше, если пробанд относится к менее поражаемому полу.
Так, в двух семьях, отягощенных язвенной болезнью желудка (рис. 32) и имеющих по двое детей разного пола, в каждой семье вероятность передачи гена одинакова для обоих детей (1/2), но вероятность заболеть больше для лиц мужского пола, так как язвенная болезнь относится к заболеваниям с преимущественным поражением лиц мужского пола. Если же сравнить такую вероятность между сыновьями двух семей, то она выше в первой семье (а), так как язвенной болезнью здесь страдает женщина, то есть в данном случае больной родитель относится к менее поражаемому полу.

Основу оценки риска при мультифакториальной патологии составляют эмпирические данные о популяционной и семейной частоте каждого заболевания или порока развития (табл. 4).
Такие таблицы составлены для большинства мультифакториальных заболеваний и пороков.
Проводя анализ родословных, можно выявить лиц, генетически предрасположенных к тому или иному заболеванию. Это позволит эффективно проводить лечебно-профилактические мероприятия, направленные на предупреждение развития у них патологии. Так, при наличии гипертонической болезни у одного из родителей необходимо с раннего возраста контролировать уровень артериального давления у детей, в дальнейшем рекомендовать им щадящий режим (избегать нервных и физических перегрузок, употребления алкоголя, курения и др.). В таких семьях необходимо правильное соблюдение режима дня, увеличение физической активности за счет умеренных физических нагрузок, аутотренинг, ограничение приема поваренной соли, регулирование массы тела и др.
Выполнение этих рекомендаций с раннего возраста окажет благотворный эффект на профилактику развития гипертонии.
Таким образом, анализ семейных данных с целью выявления групп лиц, склонных к мульфакториальным заболеваниям, - важное и перспективное направление в профилактической медицине.

МЕДИКО-ГЕНЕТИЧЕСКОЕ КОНСУЛЬТИРОВАНИЕ
ЗАДАЧИ И ОРГАНИЗАЦИЯ МЕДИКО-ГЕНЕТИЧЕСКОГО КОНСУЛЬТИРОВАНИЯ
Наиболее распространенным и эффективным методом профилактики наследственных болезней является медико-генетическое консультирование, которое представляет собой один из видов специализированной медицинской помощи населению, направленной на предупреждение появления в семье больного ребенка.
Главная цель генетического консультирования - предупреждение рождения больных детей. Это в первую очередь касается тяжелых и плохо поддающихся лечению пороков развития и наследственных болезней, приводящих к физической или психической неполноценности.
Задачами медико-генетического консультирования являются:
1) ретро- и проспективное консультирование семей и больных с наследственной или врожденной патологией;
2) пренатальная диагностика врожденных и наследственных заболеваний ультразвуковыми, цитогенетическими, биохимическими и молекулярно-генетическими методами;
3) помощь врачам различных специальностей в постановке диагноза наследственного или врожденного заболевания, если для этого требуются специальные генетические методы исследования;
4) объяснение пациенту и его семье в доступной форме о величине риска иметь больное потомство и оказание им помощи в принятии решения;
5) ведение территориального регистра семей и больных с врожденной и наследственной патологией и их диспансерное наблюдение;
6) пропаганда медико-генетических знаний среди населения.
Чаще всего за консультацией в МГЦ обращаются семьи, в которых уже есть один или несколько больных детей с наследственным или врожденным заболеванием и родителей беспокоит вопрос дальнейшего деторождения. Другая группа включает семьи, где болен один из супругов, и родителей интересует прогноз здоровья будущих детей. К третьей группе относятся семьи практически здоровых детей, у которых по линии одного или обоих родителей имеются родственники с наследственной патологией. В четвертую группу входят родители, желающие узнать, какова судьба здоровых братьев и сестер больного ребенка (не возникнет ли аналогичное заболевание у них в дальнейшем, а также у их детей).
Кроме перечисленных случаев, необходимо заподозрить наследственную патологию и направить семью в медико-генетический центр при следующих показаниях:
- наличие аналогичных заболеваний или симптомов у нескольких членов семьи;
- первичное бесплодие супругов;
- первичное невынашивание беременности;
- отставание в умственном и физическом развитии;
- рождение ребенка с врожденными пороками развития;
- первичная аменорея, особенно в сочетании с недоразвитием вторичных половых признаков;
- наличие кровного родства между супругами и др.
Особенностью работы МГЦ является то, что исследуется не только человек, обратившийся за консультацией (пробанд), но и члены его семьи. Для генетической консультации требуются подробные сведения о родственниках пробанда, часто возникает необходимость в углубленном их обследовании. Это приводит к гораздо большим затратам времени, чем на прием больного любым другим специалистом. На первичный прием семьи с составлением родословной требуется 1 ч 20 мин, на повторный приём - 30 мин.
ОСНОВНЫЕ ПРИНЦИПЫ КОНСУЛЬТИРОВАНИЯ
Консультации по прогнозу состояния здоровья потомства можно разделить на две группы: 1) проспективное консультирование; 2) ретроспективное консультирование. Наиболее эффективный вид профилактики наследственных болезней - это проспективное консультирование, то есть консультирование супружеских пар до рождения ребенка или молодых людей до вступления в брак. Такие консультации проводятся при наличии кровного родства между супругами, при неблагоприятном семейном анамнезе, при воздействии вредных средовых факторов на кого-либо из супругов (профессиональные вредности, лечебное облучение, тяжелые инфекции и др.). Ретроспективное консультирование - это консультирование семей, в которых уже имеется больной ребенок, относительно здоровья будущих детей.
Чтобы правильно рассчитать генетический риск, нужно поставить точный диагноз и определить тип наследования заболевания в данной семье. Для установления или уточнения диагноза обратившемуся за консультацией или его родственникам бывает необходимо назначить ряд инструментальных и лабораторных исследований. В связи с этим в штат медицинского персонала областных МГЦ кроме врача-генетика (педиатра) введены должности врача акушера-гинеколога, врача ультразвуковой диагностики, биохимика-генетика, врача лаборанта-цитогенетика и др.
Средний медицинский персонал (медицинские сестры, фельдшера-акушерки, фельдшера-лаборанты) обеспечивают работу врачей, участвуют во врачебном приеме, проводят забор и подготовку материала для цитогенети-ческого и биохимического исследования, ведут медицинскую документацию.
Минимальный перечень лабораторных исследований, выполняемый в областных МГЦ, включает: определение полового хроматина, кариотипирование с использованием культуры лимфоцитов периферической крови, амниоцитов, клеток хориона и плаценты, иммуноферментное определение в сыворотке крови альфа-фетопротеина и хориогонина, лотовый тест для диагностики муковисцидоза и др.
ЭТАПЫ КОНСУЛЬТИРОВАНИЯ
Медико-генетическое консультирование можно разделить на три этапа.
Первый этап - это уточнение диагноза заболевания. В ряде случаев точный диагноз заболевания может быть установлен уже перед направлением в МГЦ. Это бывает возможно в случае хорошо изученной или часто встречающейся патологии (гемофилия, сахарный диабет и др.). Однако чаще больных направляют в консультацию для установления диагноза. Так, рождение ребенка с множественными пороками развития может явиться результатом эмбрио- или фетопатии, следствием нарушения кариотипа или результатом мультифакториальной патологии. В этом случае правильный диагноз может быть поставлен только после тщательного анализа родословной, обследования пробанда и его родственников, применения цитогенетического и других специальных методов исследования.
Второй этап - определение генетического прогноза для потомства. Установив диагноз наследственного заболевания, закономерности его передачи в семье и определив, является ли данная патология следствием новой мутации или возникла как результат скрытого носительства патологической мутации на генном или хромосомном уровне, производится расчет повторного риска рождения больного ребенка в семье. Это входит в функции врача-генетика.
Генетический риск выражает вероятность появления определенной аномалии у обратившегося за консультацией или его потомков и определяется двумя способами: либо путем теоретических расчетов, основанных на генетических закономерностях, либо с помощью эмпирических данных. Некоторые принципы расчета генетического риска при моногенных и мульти-факториальных заболеваниях представлены в главе X.
При хромосомных болезнях, вызванных числовыми аномалиями хромосом, вероятность повторного рождения больного ребенка в семье крайне мала (не превышает 1 %), если известно, что ни у одного из родителей нет хромосомной аномалии, а также отсутствуют другие факторы риска (например, возраст матери). Прогноз для потомства в семье, в которой родился ребенок с транслокационной формой болезни Дауна, неблагоприятен. В таком случае необходимо определить кариотип у обоих родителей, установить, кто является носителем сбалансированной транслокации, и только после этого определить повторный риск рождения ребенка с болезнью Дауна. Так, при транслокации 14/21 риск для потомства равен 10 %, если носитель транслокации мать, и 2,5 %, если носитель отец. При транслокации 21-й хромосомы на ее гомолог риск повторного рождения больного ребенка составляет 100 % независимо от того, отец или мать несут эту транслокацию.
Прогнозирование потомства неразрывно связано с диагностикой в семьях гетерозиготных носителей патологического гена. Выявление гетерозиготных носителей особенно важно в случаях патологии с аутосомно-рецессивным типом наследования, в семьях, отягощенных заболеваниями, сцепленными с Х-хромосомой; при кровнородственных браках.
В некоторых случаях носительство можно установить уже при анализе родословной: например, если у отца женщины имеется рецессивное заболевание, сцепленное с Х-хромосомой (гемофилия, миопатия Дюшенна и др.), то такая женщина со 100 % вероятностью гетерозиготна по этому гену и риск заболевания для ее сыновей равен 50 %. При заболеваниях с аутосомно-рецессивным типом наследования родители больного и его будущие дети также являются гетерозиготными носителями данного гена. При аутосомно-доминантном наследовании с неполной пенетрантностью носителями патологического гена являются все лица, имеющие больных детей и больных родителей одновременно.
Если гетерозиготность является вероятностной на основании генеалогического анализа, можно воспользоваться клиническими и биохимическими методами. Так, повышенный уровень креатинфосфокиназы в сыворотке крови матери или сестер больного псевдогипертрофической формой миопатии Дюшенна указывает на гетерозиготность по гену миопатии. Снижение в сыворотке крови у сестер или матери больного гемофилией уровня антигемофильного глобулина может явиться вполне убедительным доказательством Наличия гетерозиготного носительства гена гемофилии. В некоторых случаях при выявлении гетерозиготных носителей патологического гена необходимо обращать внимание на «малые» клинические признаки основного заболевания.
Например, женщины-гетерозиготы по гену миопатии Дюшенна часто жалуются на небольшую слабость в ногах и быструю утомляемость; при носительсгве гена гемофилии можно выявить в анамнезе легкую или умеренную тенденцию к кровоточивости.
В настоящее время для ряда наследственных заболеваний возможна ДНК-диагностика гетерозиготных носителей патологического гена.
Если гетерозиготный носитель вступает в брак, то следует определить вероятность гетерозиготности будущего супруга (супруги) и информировать, семью о риске рождения у них больного ребенка. Гетерозиготным носителям также рекомендуется избегать родственных браков, поскольку в таких случаях увеличивается риск рождения больного ребенка.
На третьем, заключительном этапе консультирования обратившихся за
консультацией в МГЦ знакомят с генетическим прогнозом для потомства,
то есть с величиной риска рождения больного ребенка, и дают им соответствующие рекомендации.
Генетический риск до 5 % расценивается как низкий и не является противопоказанием к продолжению деторождения. Риск от 6 до 20 % принято считать средним. В этом случае рекомендации относительно планирования дальнейших беременностей зависят от тяжести наследственного или врожденного заболевания и возможности его пренатальной диагностики. Генетический риск свыше 20 % относится к категории высокого риска, и при отсутствии методов пренатальной диагностики соответствующей патологии дальнейшее деторождение в данной семье не рекомендуется.
Таким образом, давая заключение, необходимо учитывать не только величину риска рождения больного ребенка, но и тяжесть заболевания, эффективность его лечения, возможности пренатальной диагностики. Учит комбинацию этих признаков, в каждом конкретном случае дается оптимальный совет. Необходимо объяснить родителям случайность распределения генов и отсутствие их вины за рождение больного ребенка, а также сообщить им, что вероятность появления аномалии во время каждой беременности составляет 4-6 % (общепопуляционный риск). Давая свои рекомендации семье, врач не имеет права настаивать на воздержании от деторождения прерывании беременности или необходимости супругам расстаться, если они являются гетерозиготными носителями одного и того же патологического гена. Все решения по дальнейшему планированию семьи принимаются только супругами.
СОВРЕМЕННЫЕ МЕТОДЫ
ПРЕНАТАЛЬНОЙ ДИАГНОСТИКИ ВРОЖДЕННЫХ ПОРОКОВ РАЗВИТИЯ И НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ
Прогноз потомства, осуществляемый в медико-генетической консультации, является вероятностным и не позволяет ответить однозначно, завершится ли данная беременность рождением здорового или больного ребенка. Эффективность медико-генетического консультирования значительно повышается с применением современных методов дородовой диагностики, которые не только позволяют определить заболевание задолго до рождения ребенка, но и прервать беременность при поражении плода в I или II триместре. Своевременное прерывание беременности необходимо при таких наследственных болезнях и врожденных пороках, лечение которых на современном этапе не дает должных результатов или при которых изменения в организме, возникшие в период внутриутробного развития, необратимы (болезни накопления, ахондроплазия, анэнцефалия и др.).
Основными показаниями для проведения пренатальной диагностики являются:
1) наличие в семье точно установленного наследственного заболевания;
2) возраст будущей матери старше 35 лет, отца - старше 40 лет;
3) наличие у матери Х-сцепленного рецессивного патологического гена;
4) беременные, имеющие в анамнезе спонтанные аборты, мертворождения неясного генеза, детей с множественными врожденными пороками развития и с хромосомной патологией;
5) наличие структурных перестроек хромосом (особенно транслокаций и инверсий) у одного из родителей;
6) гетерозиготность обоих родителей по одной паре аллелей при аутосомно-рецессивных заболеваниях;
7) беременные из зоны повышенного радиационного фона, с тератогенным воздействием и др.
К основным методам пренатальной диагностики относятся: ультразвуковое исследование (УЗИ), амниоцентез, биопсия хориона, фетоскопия, определение альфа-фетопротеина.
Из всех методов пренатальной диагностики наиболее распространено ультразвуковое исследование плода (эхография). Метод основан на способности ультразвуковой волны отражаться от поверхности раздела двух сред, отличающихся различной плотностью, что позволяет получить их изображение на экране электронно-лучевой трубки. Это исследование проводится всем беременным женщинам трехкратно на 14-16-й, 20-21-й и 26-27-й неделях беременности. Женщин из группы риска по рождению детей с пороками развития осматривают в более ранние сроки.
С помощью ультразвукового исследования можно диагностировать грубые пороки мозга (анэнцефалию, гидроцефалию, черепно- и спинномозговые грыжи, микроцефалии); пороки конечностей (отсутствие конечности или ее части, системные скелетные дисплазии); пороки почек (агенезию или гипоплазию почек, гидронефроз, поликистоз), атрезии желудочно-кишечного тракта, пуповинные и диафрагмальные грыжи, некоторые врожденные пороки сердца.
Следует отметить, что разные формы пороков диагностируют в разные сроки беременности. Если анэнцефалия может быть распознана уже на 14-16-й неделе, то для диагностики гидронефроза необходимо исследование в значительно более поздние сроки беременности. Одни пороки развития (черепно-мозговые грыжи) могут быть выявлены уже при однократном исследовании, другие (микроцефалия, некоторые формы скелетных дисплазий) диагностируются лишь при динамическом наблюдении.
Если у плода при ультразвуковом исследовании выявляется несовместимый с жизнью порок развития, т
|
из
5.00
|
Обсуждение в статье: Порядок сбора генеалогической информации и методика составления родословной |
|
Обсуждений еще не было, будьте первым... ↓↓↓ |

Почему 1285321 студент выбрали МегаОбучалку...
Система поиска информации
Мобильная версия сайта
Удобная навигация
Нет шокирующей рекламы