 |
Главная |


Лекция №4. Термодинамика химических процессов.
|
из
5.00
|
План лекции:
- Виды термодинамических системы.
- Формы энергии и их эквивалентность.
- Первый закон термодинамики. Энтальпия. Закон Гесса.
- Второй закон термодинамики. Энтропия.
- Изобарно-изотермический потенциал (энергия Гиббса). Направление протекания химических реакций.
Понятие “термодинамическая система” означает ту часть материального мира, которая является объектом исследования. Такой системой может быть, например, стеклянная колба с раствором. Остальные объекты, находящиеся за пределами выделенной системы и оказывающие на нее влияние, называются окружением. Термодинамическая система может быть: открытой, если между системой и окружением возможен обмен веществом и энергией; закрытой, если между системой и окружением возможен обмен энергией, но невозможен обмен веществом; изолированной, когда между системой и окружением невозможен обмен ни энергией, ни веществом.
Параметры, при помощи которых описывается состояние системы, называются функциями состояния. Основным свойством любой функции состояния является независимость ее изменения от способа или, точнее, от пути изменения состояния системы.
Энергия – это мера способности системы выполнять работу. Энергия измеряется теми же единицами, что и работа. В системе СИ за единицу измерения энергии принят джоуль (Дж=1Н·1м). Наиболее известной формой энергии является тепловая энергия (теплота). Ее передача от системы 1 к системе 2 или к окружению возможна только при разности температур систем 1и 2 или системы 1 и окружения. При этом количество переданной теплоты может быть вычислено по следующей формуле:
Q = n  ∆T, (3.1)
∆T, (3.1)
где n - число молей вещества, из которого состоит система 1; 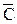 – теплоемкость этого вещества; ∆T = Т2 – Т1 , Т1 - начальная температура системы 1, Т2 - конечная температура системы 1. Теплоемкость вещества – это энергия, необходимая для повышения температуры одного моля вещества на один кельвин. Размерность
– теплоемкость этого вещества; ∆T = Т2 – Т1 , Т1 - начальная температура системы 1, Т2 - конечная температура системы 1. Теплоемкость вещества – это энергия, необходимая для повышения температуры одного моля вещества на один кельвин. Размерность 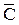 Дж/моль · К. Теплоемкость иногда относят к единице массы вещества и называют удельной теплоемкостью. Удельная теплоемкость (Суд) –это энергия, необходимая для повышения температуры одного грамма (или килограмма) вещества на один кельвин. Размерность Суд Дж/г · К (или Дж/кг · К).
Дж/моль · К. Теплоемкость иногда относят к единице массы вещества и называют удельной теплоемкостью. Удельная теплоемкость (Суд) –это энергия, необходимая для повышения температуры одного грамма (или килограмма) вещества на один кельвин. Размерность Суд Дж/г · К (или Дж/кг · К).
Механическая энергия может существовать в виде кинетической энергии и потенциальной энергии. Кинетическая энергия – это энергия, связанная с движением тела (Екин) и соотношением (3.4) определяется массой тела m и скоростью его движения v:
Eкин = (1/2)mv2. (3.4)
Потенциальная энергия – это энергия, запасенная телом. В термодинамике механическая энергия чаще всего принимает форму работы (w), которая измеряется произведением силы F на путь ее действия l, или произведением давления p на объем V. В обоих случаях размерность работы одинакова, так как давление измеряется силой, приходящейся на единицу площади:
w = Fl = pV. (3.5)
В системе СИ размерность: F – ньютон (Н), l – метр (м), p – паскаль (Па = Н/м2), V – м3, w – джоуль (Дж = Н·м). Поэтому для (3.5) будет:
Дж = Н · м = Па · м3 = (Н / м2 )м3 = Н · м. (3.6)
Потенциальная энергия частиц с зарядами q1 и q2, находящихся на расстоянии r друг от друга, определяется выражением:
Епот = q1q2 /r. (3.7)
Очевидно, что энергия химических и физических связей является одной из форм потенциальной энергии в соответствии с (3.7). Химические системы могут состоять из атомов, молекул и ионов или из любой комбинации этих частиц. Все частицы системы обладают кинетической и потенциальной энергией. Сумма кинетической и потенциальной энергии всех частиц системы называется внутренней энергией системы, которая является функцией состояния системы и обозначается буквой U. Абсолютные значения внутренней энергии систем не поддаются экспериментальному определению. Экспериментально можно только измерить изменение внутренней энергии системы:
∆U=U2-U1, (3.8)
где U1 – начальное, а U2- конечное значение внутренней энергии системы. Если U2 < U1, то ∆U имеет отрицательное значение (∆U < 0). Это происходит в том случае, когда система теряет энергию, отдавая ее другой системе или окружению, как по (3.1).
Существует еще один способ передачи энергии от одной системы другой системе – это выполнение системой или над системой работы. В химии чаще всего приходится иметь дело с работой расширения или сжатия системы (изменения объема). Например, если в химической реакции выделяется газ, то работа, выполняемая в этом случае реакционной системой, определяется выражением:
w = -p ∆V, (3.10)
где p – внешнее давление (например, атмосферное), ∆V – изменение объема системы. Знак минус в (3.10) указывает на то, что работа выполняется системой, а следовательно, система отдает энергию.
Экспериментально установленная эквивалентность различных форм энергии (как, например, (3.3)) явилась основой для вывода о том, что энергия сохраняется во всех физических и химических превращениях. Этот вывод называется первым законом термодинамики и для закрытых систем математически выражается уравнением:
∆U=Q+w, (3.11)
где каждая величина может иметь положительное или отрицательное значение. Так, если Q>0, то система приобретает тепловую энергию, если Q<0, то система теряет тепловую энергию. Аналогично, если работа совершается над системой, то w>0, а если система совершает работу, то w<0. При этом ∆U также может быть положительной и отрицательной величиной. Все три величины уравнения (3.11) измеряются в единицах энергии – джоулях.
Итак, первый закон термодинамики устанавливает, что внутренняя энергия системы может изменяться – убывать или возрастать за счет отдачи или приобретения теплоты и совершения системой или над системой работы. Так как эта формулировка отражает различные проявления закона сохранения энергии, то первый закон термодинамики иногда формулируют следующим образом. Энергию можно превратить из одной формы в другую, но ее нельзя создать или уничтожить и общее количество энергии в изолированной системе постоянно.
В некоторых химических процессах реагенты и продукты могут быть только твердыми и жидкими. Тогда при Δn=0 (Δn = n(продуктов) – n(реагентов)) изменением объема химических систем в результате реакций либо можно пренебречь (∆V=0), либо оно в точности равно нулю. В этих случаях работа по изменению объема систем тоже равна нулю w=0. Поэтому теплота, выделяемая или поглощаемая, полностью идет на изменение внутренней энергии систем – то есть одной из термодинамических функций:
∆U = Q(v), (3.15)
где индекс (v) у Q указывает, что это теплота, поглощаемая или выделяемая при постоянном объеме.
Однако, чаще всего химические реакции проводятся в открытых емкостях (например, колбах, пробирках и т. д.) и с изменением агрегатных состояний веществ (например, с участием веществ в газообразных и конденсированных состояниях). В ходе таких реакций давление в системах совпадает с атмосферным давлением, а изменение объема систем не равно нулю. В соответствии с (3.14) и при условии увеличения в ходе реакций объема систем выполнение первого закона термодинамики для них можно представить в виде (3.16):
∆U = Q(p) - p∆V, (3.16)
где индекс (p) у Q указывает на то, что это теплота, поглощаемая или выделяемая при постоянном давлении. Тепловой эффект Q(p) из (3.16) можно выразить следующим образом:
Q(p) = ∆U + p ∆V . (3.17)
По аналогии с (3.15) теплота Q(p) полностью идет на изменение термодинамической функции систем, которую назвали энтальпией (от греческого слова энтальпо – нагреваю) и обозначили буквой Н. Поэтому вместо (3.17) можно написать (3.18) при р = соnst:
Q(p) = ∆H. (3.18)
Если в (3.17) величины ∆U и ∆V выразить соответственно как (U2-U1) и (V2-V1), то получим следующее выражение:
Q(p) = U2-U1 + p(V2-V1) = ( U2+p V2)- (U1+p V1) = Н2-Н1. (3.19)
По (3.19) видно, что при переходе системы от состояния со значением функции Н1 к состоянию со значением функции Н2 система приняла количество теплоты Q(p), то есть ее теплосодержание возросло. Поэтому термодинамическую функцию Н называют также теплосодержанием. Из (3.19) следует выражение для Н в виде (3.20):
Н =U + p V (при р = соnst). (3.20)
Второе слагаемое (3.20) называют иногда внешней энергией. Так как по (3.20) Н содержит в себе внутреннюю энергию U, то абсолютную величину Н определить невозможно (как и U). Однако изменение Н в результате перехода системы от одного состояния к другому состоянию определить можно по (3.18):
Q(p) = Н2-Н1 = ∆H.
Для химических реакций изменение энтальпии – это их тепловые эффекты, учитывающие изменение объема реакционных систем.
В термохимии принято считать стандартные энтальпии образования химических элементов и элементарных веществ в наиболее устойчивой форме равными нулю. Например, энтальпия образования графита, как наиболее устойчивой формы углерода при стандартных условиях, равна нулю. Для алмаза и газообразного углерода аналогичные энтальпии отличаются от нуля:
∆Hº(C(граф))=0; ∆Hº(C(алмаз))=1,88 кДж/моль;
∆Hº(C(г)) = 718,4 кДж/моль. (3.25)
Из этого примера видно, что необходимо учитывать в каком агрегатном и фазовом состояниях находятся реагенты и продукты реакций. Поэтому в уравнениях реакций (как показано и в (3.24)) необходимо отмечать в скобках состояние вещества: (г) – газообразное; (ж) – жидкое; (к) – кристаллическое; (тв) – твердое.
Рассмотрим далее реакцию сгорания графита:
C(граф) + О2(г) = СО2(г) +393,5кДж . (3.26)
Это термохимическая форма записи уравнения реакции. В термодинамике тепловые эффекты в виде энтальпийных эффектов указываются рядом с уравнениями реакций. Вместо (3.26) можно написать следующее уравнение:
C(граф) + О2(г) = СО2(г); ∆Hр = -393,5кДж. (3.27)
Для такой формы записи уравнений, как (3.26), ∆Hºр = - Qºp.
Учитывая закон сохранения энергии (в данном случае энтальпии), для уравнения (3.26) напишем следующее равенство:
 - О + О = ∆Hºр(СО2(г)) + 393,5кДж, (3.28)
- О + О = ∆Hºр(СО2(г)) + 393,5кДж, (3.28)
где нули – это стандартные энтальпии образования элементарных веществ: графита и кислорода. Из (3.28) следует:
 ∆Hº(СО2(г)) = -393,5 кДж/моль. (3.29)
∆Hº(СО2(г)) = -393,5 кДж/моль. (3.29)
Полученная величина названа стандартной энтальпией образования оксида углерода IV (химического соединения) из элементарных веществ. Стандартные энтальпии образования химических соединений из элементарных веществ в их стандартных состояниях при температуре 298К и давлении 1 атм не равны нулю и приводятся в различных таблицах термодинамических функций.
Анализируя уравнения (3.26) - (3.29), можно показать, что для уравнений любых химических реакций выполняется уравнение (3.30):
∆Hºр =Σ∆Hº(продуктов) - Σ∆Hº(реагентов) . (3.30)
Суммы энтальпий должны включать стехиометрические коэффициенты уравнений реакций в виде множителей соответствующих значений стандартных энтальпий образования веществ.
Оксид углерода IV, полученный по реакции (3.26), можно разложить на элементарные вещества: графит и кислород – в соответствии с первым законом термохимии, который называют также законом Лавуазье и Лапласа. Если при образовании какого-либо химического соединения из элементарных веществ выделяется определенное количество теплоты, то такое же количество теплоты требуется затратить на разложение этого химического соединения на элементарные вещества. Авторы этого закона французские ученые А. Лавуазье и П. Лаплас (1870 г.).
СО2(г) = С(граф) + О2(г) . (3.33)
Так как по (3.29) ∆Hº (СО2(г)) = -393,5 кДж/моль, то по (3.30) вычислим ∆Hºр (3.33):
∆Hºр = О +О -1 моль · ∆Hº (СО2(г)) = 393,5 кДж.
Положительное значение ∆Hºр указывает на то, что реакция (3.33) эндотермическая, а величина ∆Hºр соответствует выводу первого закона термохимии (∆Hºр (3.33) = - ∆Hºр(3.26)).
Второй закон термохимии или закон Гесса имеет следующую формулировку: суммарный тепловой эффект химической реакции при постоянном давлении не зависит от ее промежуточных стадий. Этот закон можно сформулировать короче: энтальпийный эффект реакции не зависит от промежуточных стадий. Автор закона Г. И. Гесс, немец по происхождению, работавший профессором Петербургского горного института и установивший этот закон в 1840 г.
Закон Гесса применяется в тех случаях, когда химические превращения могут быть осуществлены несколькими различными путями. При этом результирующее изменение энтальпии одинаково для любого пути. Например, сгорание графита до оксида углерода IV может быть осуществлено двумя путями (а) и (б). Первый путь – это реакция (а) в одну стадию, а второй путь – это процесс в две стадии (б1) и (б2):
(а) C(граф) + O2(г)=СО2(г); ∆Hºр (а),
(б1) C(граф) + 0,5O2(г) = СО(г); ∆Hºр(б1),
(б2) СО(г) + 0,5О2(г) = СО2(г); ∆Hºр(б2). (3.34)
∆Hºр(а) = ∆Hºр(б1) + ∆Hºр(б2)
Рассмотренное применение закона Гесса к двум путям получения оксида углерода IV может быть представлено либо в виде схемы – рисунка 3.3.

Рис. 3.3. Иллюстрация к закону Гесса:
Обратимым называется процесс, который в любой момент времени можно заставить протекать в обратном направлении, изменив какую-нибудь независимую переменную на бесконечно малую величину. Рассмотрим в качестве примера процесс теплообмена между двумя закрытыми системами I и II (рис.3.5). В нем независимой переменной является температура. Ее бесконечно малое изменение можно обозначить следующим образом:
dT = T2 - T1 , (3.35)
где d – называется дифференциалом (латинское слово, обозначающее разность), а dT – дифференциал температуры, показывающий, что T1 и T2 различаются между собой на бесконечно малую величину.

Рис. 3.5. Схема теплообмена между закрытыми системами I и II:
1- емкость с водой и льдом; 2- лед; 3 – вода;
4 – тепловая изоляция; 5 – пробное тело;
6 – емкость с теплоносителем
Например, для системы I при Т = 273,15К будет выполняться следующее уравнение:
 H2O(к Н2О(ж). (3.36)
H2O(к Н2О(ж). (3.36)
Система II представляет собой тепловой резервуар, теплоносителем которого могут быть различные вещества, включая воду. Пробное тело (5) – это небольшой металлический шарик, укрепленный на ручке, изготовленной из полимерного материала, плохо проводящего теплоту.
Если температура теплоносителя системы II будет незначительно выше 273,15К, то, касаясь пробным телом неизолированных участков вначале системы II, а затем системы I, мы перенесем от системы II системе I порцию теплоты dQ. Эту операцию можно повторять многократно. Каждая порция теплоты dQ будет увеличивать на бесконечно малую величину температуру воды в емкости (1) (на dT), что будет приводить к плавлению бесконечно малой порции льда и образованию из нее такой же бесконечно малой порции воды dn. В последней будет сосредотачиваться (запасаться) теплота dQ в виде скрытой теплоты плавления:
dQ =  Hºплdn . (3.37)
Hºплdn . (3.37)
Для того чтобы расплавилось заметное количество льда (например, заштрихованная часть куска льда на рис. 3.5) и образовалось такое же количество воды, описанный процесс теплообмена порциями dQ необходимо проводить бесконечно долго (назовем его прямым процессом). При этом все количество переданной от системы II системе I теплоты можно вычислить суммированием бесконечно малых порций dQ. Такое суммирование называется интегрированием и обозначается растянутой латинской буквой S. В нашем случае это будет следующее выражение:
 (3.38)
(3.38)
Для конкретного решения подобных задач необходимо знать пределы интегрирования, которые указывают в виде верхнего и нижнего индексов знака интеграла. Так, для числовой оси значений x интеграл и его решение в пределах от x 1 до x 2 будут следующими:
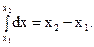 (3.39)
(3.39)
Обозначим начальное и конечное количества воды в системе I соответственно n1и n2. Тогда, с учетом (3.37)-(3.39) и того, что постоянные величины выносятся из под знака интегрирования, получим:
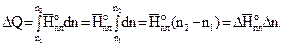 (3.40)
(3.40)
Только в обратимом процессе запасается такое количество теплоты, которого достаточно для протекания обратного процесса и возвращения системы к начальному состоянию. Рассматриваемый прямой процесс можно было в любой момент времени повернуть в обратное направление. Мы это сделаем после завершения прямого процесса.
Допустим, что, понизив предварительно температуру теплоносителя системы II до значения, незначительно ниже 273,15 К, мы пробным телом будем касаться вначале системы I, затем системы II. При этом теплота бесконечно малыми порциями dQ будет передаваться от системы I системе II. Если такой обратный процесс проводить бесконечно долго, то часть воды в системе I закристаллизуется и, если количествo образовавшегося при этом льда будет равно ∆n, то система I передаст системе II такое же количество теплоты ∆Q, как в прямом процессе:
 (3.41)
(3.41)
где штрихом помечены числа молей льда. Для системы I в обратимом процессе выполнится равенство:
∆Q(прям.) = -∆Q(обрат.), (3.42)
а после завершения всего цикла она придет к исходному состоянию с количествами воды и льда соответственно а и в при Т = 273,15К.
Если рассмотренные прямой и обратный процессы проводить с заметной скоростью за какой-то конечный промежуток времени, то теплоту от одной системы к другой системе необходимо передавать большими количествами. При этом будут неизбежны потери теплоты в окружение и не выполнится равенство (3.42):
∆Q(прям.) ≠ -∆Q(обрат.). (3.43)
В этом случае процесс будет необратимым.
В качестве примера можно рассмотреть реакцию сгорания пропана (бытового газа) в газовой горелке:
С3Н8(г)+5О2(г) = 3СО2(г)+4Н2О(г); ∆Нºр = -2202 кДж. (3.44)
Уменьшение потенциальной энергии реакционной системы (3.44) равно:
∆Hºр = ∆Hºпрод - ∆Hºреаг = -2202 кДж.
Самопроизвольным процессом является и реакция сгорания графита (3.26), ∆Hºр которой отрицательно. О реакциях, которые протекают самопроизвольно и с выделением энергии (экзотермические реакции), говорят, что их движущим фактором является энтальпийный фактор (∆Hºр<0).
Наряду с самопроизвольными экзотермическими процессами известны самопроизвольные эндотермические процессы. Например: плавление льда при стандартных условиях –
Н2О (к) → Н2О (ж); ∆Нº = 6,0 кДж/моль ;
растворение хлорида калия в воде –
КСl(к) + Вода → КСl(водн); ∆Нº = 19 кДж/моль;
испарение воды –
Н2О (ж) → Н2О (г); ∆Нº = 44 кДж/моль.
Все три примера имеют одно общее свойство – частицы исходных состояний более упорядочены, чем частицы конечных состояний. Действительно, регулярная структура льда из молекул воды в узлах кристаллической решетки при плавлении заменяется жидкими ассоциатами молекул воды в среде жидкой воды, не имеющей регулярного строения. Растворение хлорида калия также сопровождается разрушением кристаллической решетки и возникновением беспорядочного распределения ионов К+ и Сl¯ в воде. Когда испаряется вода, ассоциаты из ее молекул заменяются отдельными молекулами, движущимися независимо друг от друга в газовой среде. Следовательно, стремление к увеличению неупорядоченности является движущим фактором самопроизвольных эндотермических процессов.
Так как изменение величин энтальпии ∆Н (и внутренней энергии ∆U) не дает однозначного ответа на вопрос о возможности самопроизвольного протекания химических реакций и физических процессов, то для этой цели введена еще одна термодинамическая функция, названная энтропией. Энтропия – это термодинамическая функция, характеризующая тепловое состояние систем, а также меру их неупорядоченности. Ее обозначают латинской прописной буквой S. Дифференциал энтропии (бесконечно малое изменение энтропии) выражается следующей формулой:
dS = dQ/T . (3.45)
Учитывая формулы (3.40) и (3.45), можно показать, как изменяется энтропия одного моля кристаллического вещества (состояние I) вследствие обратимого перехода его в жидкое состояние (состояние II) при температуре плавления:
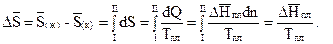 (3.46)
(3.46)
Так как ∆Hпл > 0, то ∆S > 0, а это значит, что при одной и той же температуре энтропия жидкости больше энтропии кристаллического тела при их одинаковых количествах (n(к) =n(ж)). Иными словами, при фазовом переходе кристалла в жидкое состояние энтропия возрастает. Возрастает и неупорядоченность частиц вещества.
Аналогично при температуре кипения обратимый переход одного моля жидкости в парообразное состояние также сопровождается возрастанием энтропии:
 (3.47)
(3.47)
А обратимое нагревание одного моля вещества в любом агрегатном состоянии без фазовых переходов с учетом (3.45) и в соответствии с известным табличным решением интеграла  приводит к возрастанию энтропии:
приводит к возрастанию энтропии:
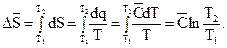 (3.48)
(3.48)
При Т2>Т1 ln(T2/T1)>0 и, следовательно,  >0. То, что энтропия всегда больше при более высокой температуре, согласуется с большей неупорядоченностью теплового движения структурных частиц при более высокой температуре. На основании уравнений (3.46) – (3.48) для одинаковых количеств вещества, в частности при n=1, выполняется следующее неравенство:
>0. То, что энтропия всегда больше при более высокой температуре, согласуется с большей неупорядоченностью теплового движения структурных частиц при более высокой температуре. На основании уравнений (3.46) – (3.48) для одинаковых количеств вещества, в частности при n=1, выполняется следующее неравенство:
S(тв) < S(ж) < S(г). (3.49)
Значения энтропии различных веществ при нахождении их в стандартных условиях даны в таблицах. Поэтому изменение энтропии химических систем в результате реакций, а также систем и веществ в результате фазовых переходов можно вычислить. Например, в реакции сгорания одного моля водорода:
Н2(г) + 0,5 О2(г) = Н2О(г).
 Sº, Дж/(моль·K) 131,0 205,0 189,0 (3.50)
Sº, Дж/(моль·K) 131,0 205,0 189,0 (3.50)
Изменение энтропии в химической реакции определяется следующим уравнением:
∆Sºp=Σ Sº (продуктов) - Σ Sº (реагентов), (3.51)
где в суммы Sº(продуктов) и Sº(реагентов) входят соответствующие сомножители, равные стехиометрическим коэффициентам веществ в уравнении реакции. Для реакции (3.50) по (3.51) получим:
∆Sºp = [1моль · 189 Дж/(моль·К)] - [0,5 моль · 205 Дж/(моль·К)]-
- [1моль· 131 Дж/(моль·К)] = -44,5Дж/К.
Полученный результат показывает, что энтропия реакционной системы уменьшилась (∆Sºp <0).
Второй закон термодинамики рассматривается как постулат, справедливость которого доказывается результатами его применения. Существует много формулировок этого закона, но все они приводят к одним и тем же результатам. Их главная мысль в том, что в любой изолированной системе с течением времени происходит постоянное возрастание беспорядка, т.е. энтропии. Второй закон термодинамики утверждает, что все самопроизвольные процессы протекают обязательно с увеличением суммарной энтропии системы и ее окружения:
ΔSсум > 0; ∆Sсум = ∆Sсист + ∆Sокруж , (3.53)
где ∆Sсум – суммарное изменение энтропии системы (∆Sсист) и окружения (∆Sокруж).
Реакция сгорания водорода (3.50) после инициирования (под-жигания) протекает самопроизвольно, следовательно, по второму закону термодинамики полное изменение энтропии ∆Sсум должно быть больше нуля. Результат, полученный по (3.51) для этой реакции ∆Sºр= – 44,5Дж/К, является неполным и относится только к системе, т.е. это ∆Sсист. Для вычисления ∆Sсум надо еще определить величину изменения энтропии окружения ∆Sокруж. Так как реакция (3.50) экзотермическая, то пользуясь табличными стандартными значениями энтальпии образования веществ, вычислим тепловой эффект этой реакции в виде ∆Нºр:
Н2(г) + 0,5О2(г) = Н2О(г);
 ∆Hº, кДж/моль 0 0 -285,8
∆Hº, кДж/моль 0 0 -285,8
∆Нºр = 1моль·(-285,8 кДж/моль) = -285,8 кДж.
Теплота ∆Нºр выделяется и рассеивается в окружении реакционной системы, энтропия которого должна возрасти на ∆Sокруж:
∆Sºокруж = - ∆Нºр/Тº. (3.54)
Тогда для реакции (3.50) получим:
∆Sºсум = ∆Sºр - ∆Нºр/Тº == - 44,5 Дж/К- (-285,8·103Дж/298К) = +914,6 Дж/К,
∆Sºсум > 0. (3.55)
Этот результат удовлетворяет условие второго закона термодинамики.
В рассмотренных примерах параграфа 3.5 показано, что известны два движущих фактора самопроизвольных процессов: энтальпийный (∆Н<0) и энтропийный (∆S>0). Для учета обоих движущих факторов самопроизвольных процессов введена термодинамическая функция, названная свободной энергией или энергией Гиббса. Ее также называют изобарным или изобарно-изотермическим потенциалом. Энергия Гиббса обозначается буквой G. Уравнение для вычисления G или ∆G может быть выведено несколькими способами, например, с использованием общего выражения уравнения (3.55) для самопроизвольного процесса:
∆Sсум = ∆Sсист - ∆Нсист /Т .
Для этого умножим обе части уравнения на – Т:
-Т∆Sсум = ∆Нсист - Т∆Sсист . (3.56)
В полученном уравнении ΔНсист – это изменение всей энергии системы, -ТΔSсист – это изменение связанной энергии, которая идет на изменение энтропии системы. В левой части равенства величина -ТΔSсист определяет изменение свободной энергии реакционной системы, которую можно использовать на выполнение нужной работы (т.е. это энергия доступная для использования):
ΔG = -ТΔSсум. (3.57)
Равенство (3.57) позволяет записать уравнение (3.56) в следующем виде:
ΔG = ΔН - ТΔS, (3.58)
где все величины ∆G, ΔН и ∆S относятся к реакционной системе. По (3.58) видно, что ∆G учитывает изменение энтальпии и энтропии, т.е. оба движущие фактора самопроизвольных процессов. При этом ∆G самопроизвольных процессов должно быть меньше нуля, величиной отрицательной. Действительно, в правой части равенства (3.57) Т всегда больше нуля, а ∆Sсум больше нуля по второму закону термодинамики, следовательно, для самопроизвольного процесса ΔG<0.
По аналогии со стандартной энтальпией образования химического соединения, введено понятие о стандартной свободной энергии образования химического соединения. Стандартной свободной энергией образования (  ) называется изменение свободной энергии, которым сопровождается образование при стандартных условиях одного моля химического соединения из входящих в него элементов в их стандартных состояниях. Стандартные свободные энергии образования элементов в их стандартных состояниях равны нулю. Величины
) называется изменение свободной энергии, которым сопровождается образование при стандартных условиях одного моля химического соединения из входящих в него элементов в их стандартных состояниях. Стандартные свободные энергии образования элементов в их стандартных состояниях равны нулю. Величины  химических соединений даны в таблицах (например, приложение 1).
химических соединений даны в таблицах (например, приложение 1).
Стандартное изменение свободной энергии химической реакции (∆Gºp) можно вычислить двумя способами. Во-первых, его можно вычислить по табличным величинам свободных энергий образования продуктов и реагентов с использованием следующего уравнения:
∆Gºp=Σ∆Gº(продуктов) - Σ∆Gº(реагентов) (3.59)
и с учетом стехиометрических коэффициентов уравнения химической реакции. Например:
СО(г) + 0,5 О2(г) = СО2 (г)
∆Gº, кДж/моль -137,1 0 -394,4
∆Gºр = 1 моль·(-394,4кДж/моль) - 0,5 моль·(0 кДж/моль) -
- 1моль · (-137,4 кДж/моль) = - 257,3кДж.
Во-вторых, стандартное изменение свободной энергии химической реакции можно вычислить по стандартным изменениям энтальпии (∆Нºp) и энтропии (∆Sºp) рассматриваемой реакции с помощью уравнения (3.58).
Лекция №5.
Растворы.
План лекции:
- Дисперсные системы, их классификация.
- Способы выражения концентрации растворов. Растворимость.
- Электролитическая диссоциация.
- Ионное произведение воды. Водородный показатель.
- Ионнообменные реакции. Гидролиз солей.
- Смещение равновесия при растворении. Индикаторы.
Фаза – это совокупность частей физико-химической системы, обладающих одинаковыми физическими и химическими свойствами. Гетерогенные дисперсные системы содержат несколько фаз (по крайней мере две), равномерно распределенных друг в друге. Из них различают непрерывную фазу, называемую дисперсионной средой, и раздробленную, дискретную, называемую дисперсной фазой. Чаще всего фазы состоят из различных веществ, т.е. большинство гетерогенных дисперсных систем являются многокомпонентными, например, цементный раствор, состоящий из дисперсионной среды – воды и твердой дисперсной фазы – частиц цементного клинкера. Очевидными признаками гетерогенных дисперсных систем также являются наличие в них границ раздела фаз и непостоянство состава (его можно произвольно изменять в некоторых пределах).
Гетерогенные дисперсные системы классифицируют по двум признакам. Во-первых, в зависимости от размера частиц дисперсной фазы (d) они разделяются на грубодисперсные (d > 10-7 м) и коллоидные (d от 10-7 до 10-9 м). Во-вторых, в зависимости от агрегатного состояния фаз различают пены (газообразная дисперсная фаза в жидкой или твердой дисперсионной среде), эмульсии (жидкая дисперсная фаза в жидкой или твердой дисперсионной среде) и золи (твердая дисперсная фаза в жидкой, газообразной или твердой дисперсионной среде).
Растворы – это гомогенные многокомпонентные системы, не имеющие строго определенного состава (его, как и состав гетерогенных дисперсных систем, можно изменять, иногда в очень широких пределах). В них нет отдельных различных фаз и нет границ раздела. Размер частиц растворенных веществ соизмерим с размером структурных частиц веществ - молекул, ионов, атомов (d < 10-9 м). Если один из компонентов раствора находится в большем количестве, чем другие, то его называют растворителем. Другие компоненты в этом случае называют растворенными веществами. Растворителем считают также то вещество, агрегатное состояние которого при образовании раствора не изменяется. В зависимости от агрегатного состояния растворителя растворы бывают жидкими, газообразными и твердыми.
1. Молярная концентрация (с) показывает, сколько молей растворенного вещества содержится в 1 литре раствора:
с = n1/V3 = m1/M1V3, моль/л. (5.8)
2. Эквивалентная концентрация или нормальность (сэ) показывает, сколько молей эквивалентов вещества (nэ) содержится в 1 литре раствора:
сэ = nэ(1) / V3 = m1 / Mэ(1) V3, моль/л. (5.9)
3. Моляльная концентрация, или моляльность, (b) показывает, сколько молей вещества содержится в 1 кг растворителя:
b = n1 / m2 = m1 / M1m2, моль/кг растворителя. (5.10)
4. Мольная доля (хi) – это отношение числа молей одного из компонентов раствора к сумме молей веществ, входящих в раствор. Для бинарного раствора х1 + х2 = 1.
x1 = n1 / (n1 + n2), x2 = n2 / (n1 + n2), (5.11)
х1- мольная доля растворенного вещества; х2 - мольная доля растворителя.
5. Массовая (ωi) или объемная (φi) доли одного из компонентов раствора являются безразмерными величинами, равными отношению массы или объема растворенного вещества к общей массе или к общему объему раствора:
ω1 = m1 / m3, φ1 = V1 / V3. (5.12)
6. Титр раствора (Т) – это число граммов растворенного вещества в 1 см3 (1 мл) раствора. Он определяется отношением массы растворенного вещества (m1 в г) к объему раствора (V3 в мл):
Т = m1 / V3, г / мл. (5.13)
Опытным путем установлено, что вещества лучше растворяются в тех растворителях, которые химически им подобны. Это обобщение часто формулируется в виде более простой фразы: подобное <
|
из
5.00
|
Обсуждение в статье: Лекция №4. Термодинамика химических процессов. |
|
Обсуждений еще не было, будьте первым... ↓↓↓ |

Почему 1285321 студент выбрали МегаОбучалку...
Система поиска информации
Мобильная версия сайта
Удобная навигация
Нет шокирующей рекламы